

Glucagon Receptor Signaling and Lipid Metabolism
- PMID: 31068828
- PMCID: PMC6491692
- DOI: 10.3389/fphys.2019.00413
Glucagon Receptor Signaling and Lipid Metabolism
Abstract
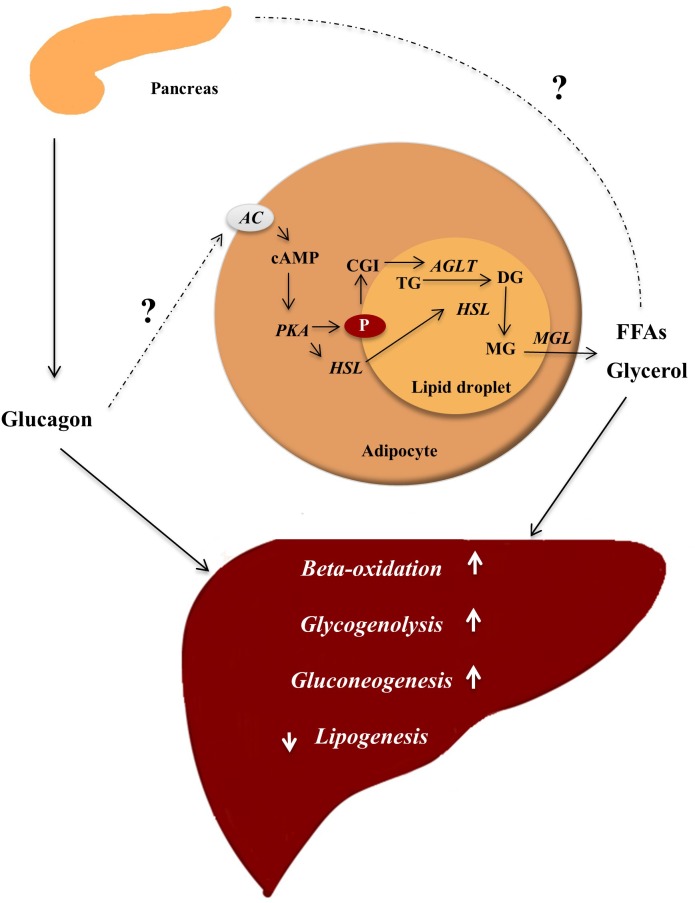
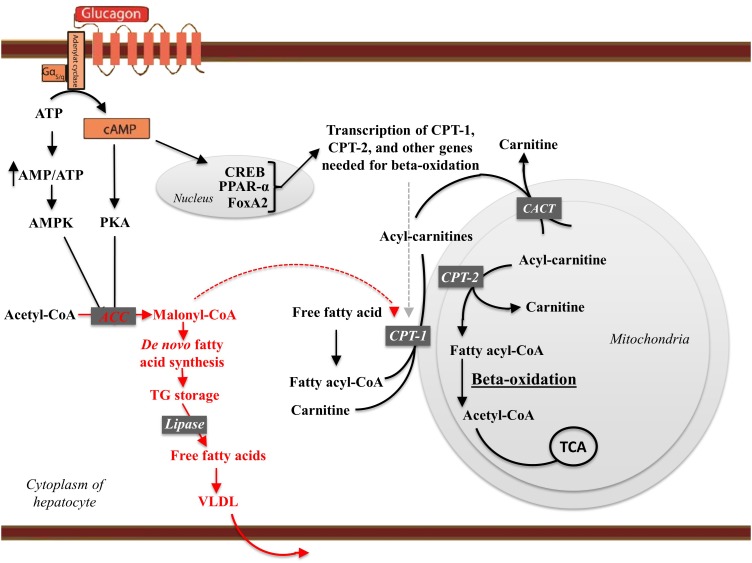
Glucagon is secreted from the pancreatic alpha cells upon hypoglycemia and stimulates hepatic glucose production. Type 2 diabetes is associated with dysregulated glucagon secretion, and increased glucagon concentrations contribute to the diabetic hyperglycemia. Antagonists of the glucagon receptor have been considered as glucose-lowering therapy in type 2 diabetes patients, but their clinical applicability has been questioned because of reports of therapy-induced increments in liver fat content and increased plasma concentrations of low-density lipoprotein. Conversely, in animal models, increased glucagon receptor signaling has been linked to improved lipid metabolism. Glucagon acts primarily on the liver and by regulating hepatic lipid metabolism glucagon may reduce hepatic lipid accumulation and decrease hepatic lipid secretion. Regarding whole-body lipid metabolism, it is controversial to what extent glucagon influences lipolysis in adipose tissue, particularly in humans. Glucagon receptor agonists combined with glucagon-like peptide 1 receptor agonists (dual agonists) improve dyslipidemia and reduce hepatic steatosis. Collectively, emerging data support an essential role of glucagon for lipid metabolism.
Keywords: adipose tissue; alpha cell; glucagon; lipid; liver.
Figures
References
-
- Adriaenssens A. E., Svendsen B., Lam B. Y., Yeo G. S., Holst J. J., Reimann F., et al. (2016). Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59 2156–2165. 10.1007/s00125-016-4033-1 - DOI - PMC - PubMed
-
- Anthonsen M. W., Ronnstrand L., Wernstedt C., Degerman E., Holm C. (1998). Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 273 215–221. 10.1074/jbc.273.1.215 - DOI - PubMed
LinkOut - more resources
Full Text Sources