

Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development
- PMID: 31072004
- PMCID: PMC6540044
- DOI: 10.3390/ijms20092275
Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development
Abstract
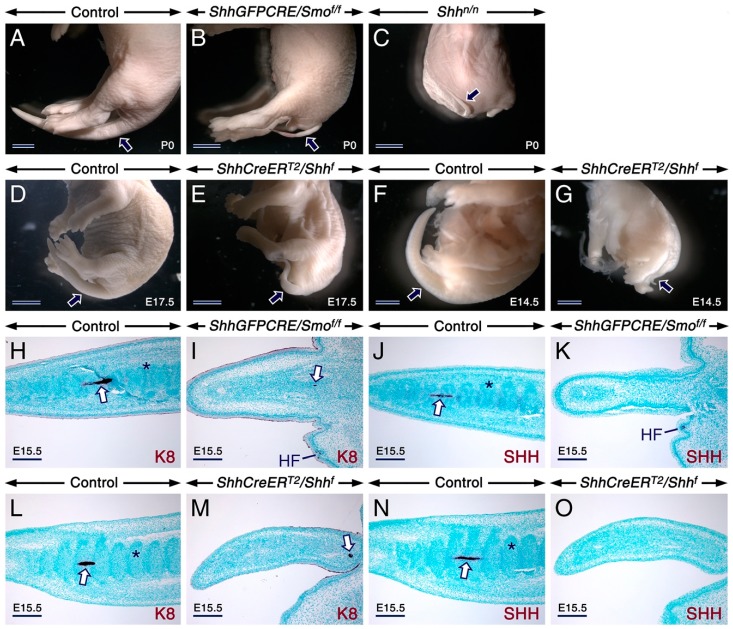
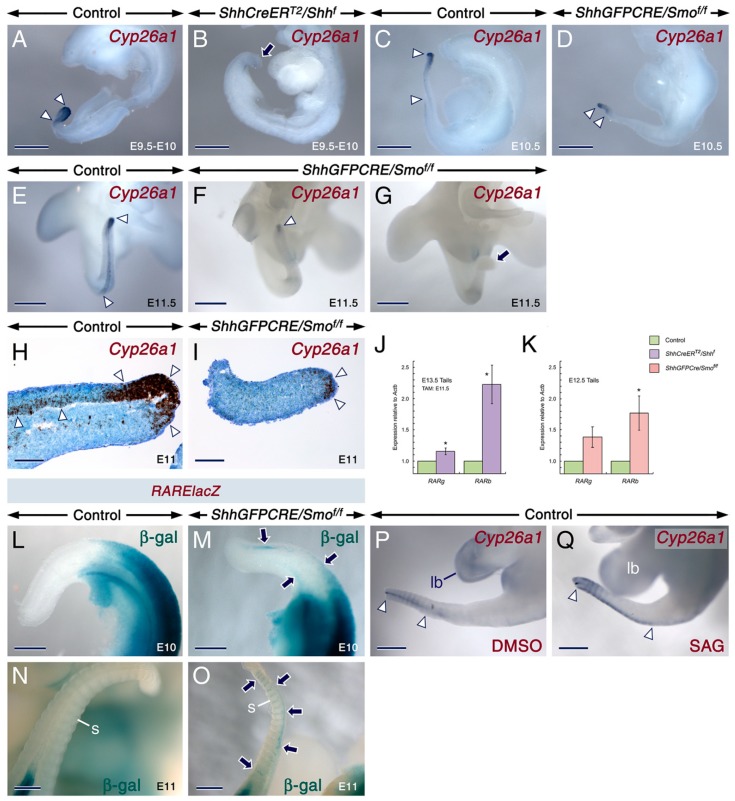
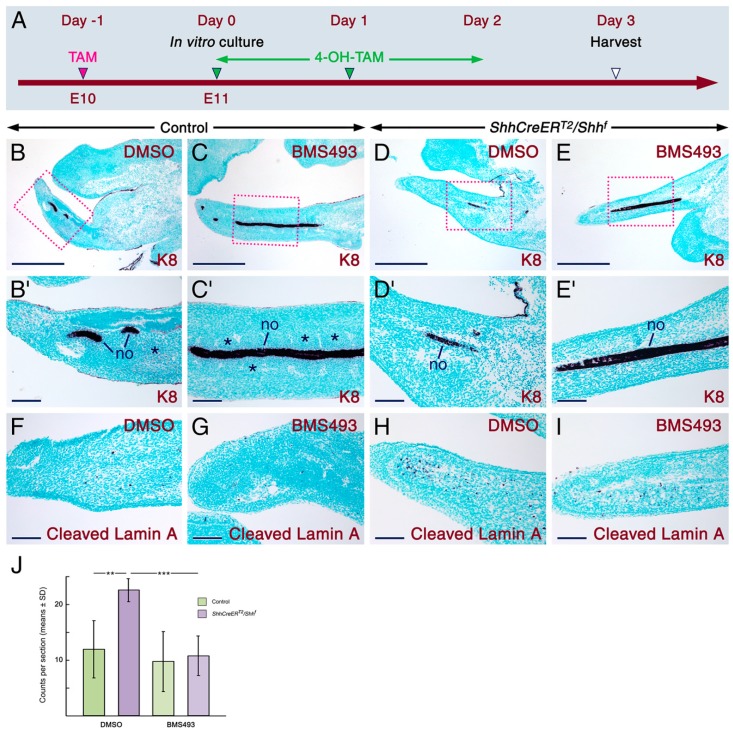
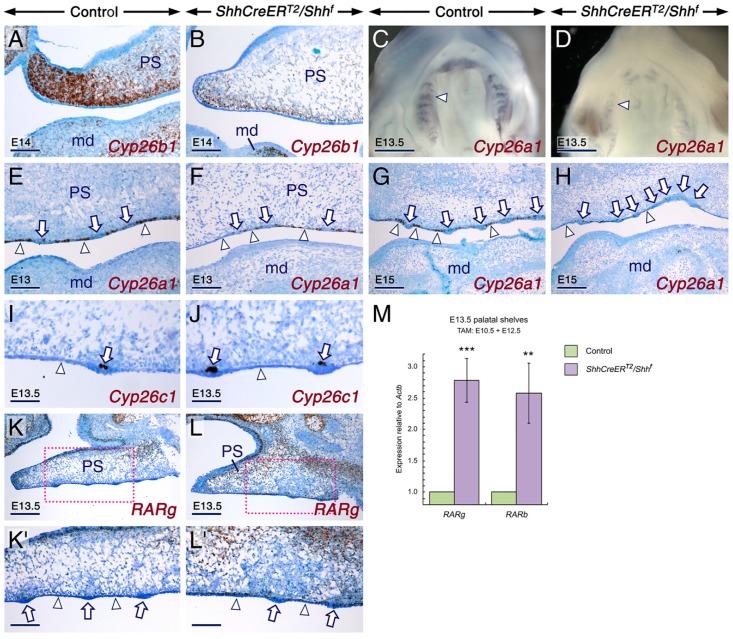
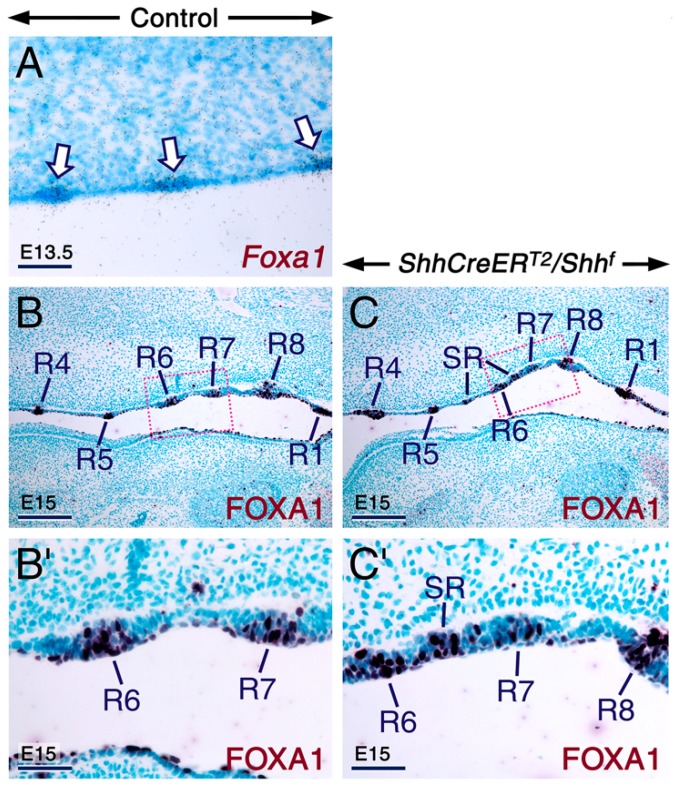
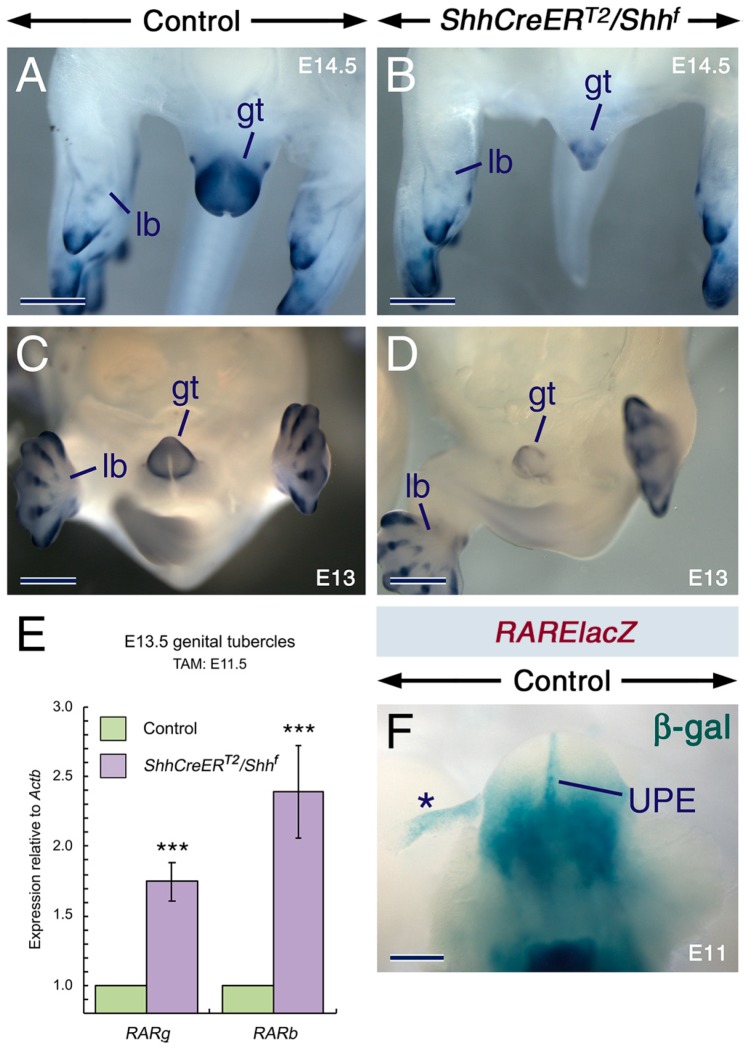
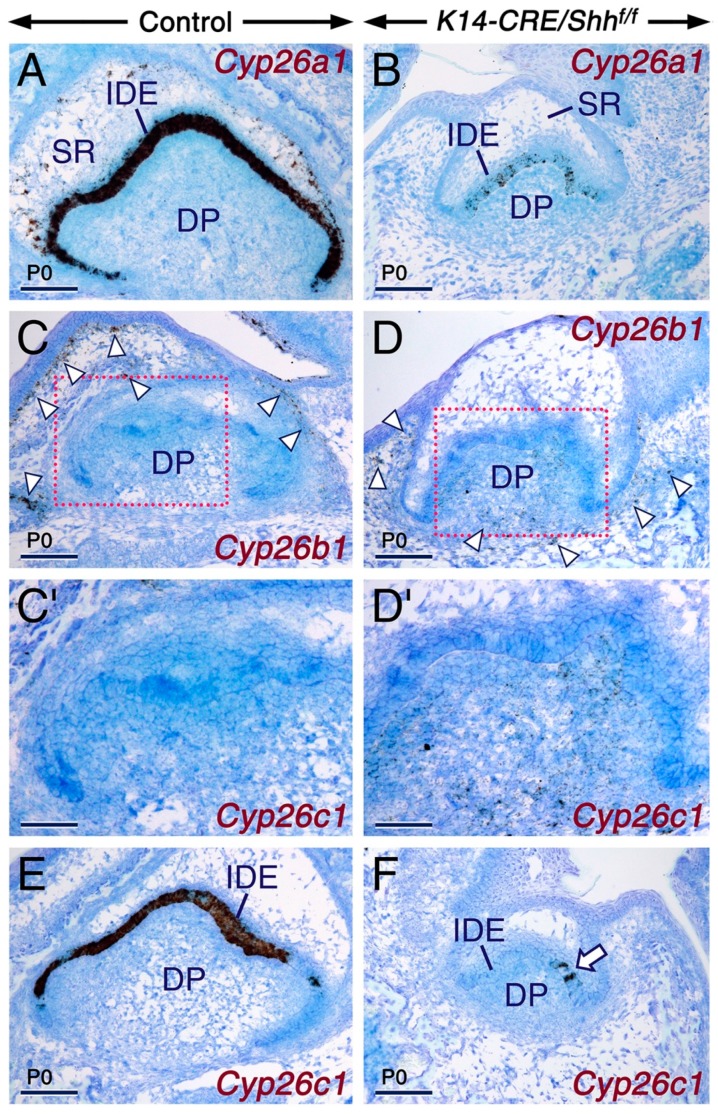
Deciphering how signaling pathways interact during development is necessary for understanding the etiopathogenesis of congenital malformations and disease. In several embryonic structures, components of the Hedgehog and retinoic acid pathways, two potent players in development and disease are expressed and operate in the same or adjacent tissues and cells. Yet whether and, if so, how these pathways interact during organogenesis is, to a large extent, unclear. Using genetic and experimental approaches in the mouse, we show that during development of ontogenetically different organs, including the tail, genital tubercle, and secondary palate, Sonic hedgehog (SHH) loss-of-function causes anomalies phenocopying those induced by enhanced retinoic acid signaling and that SHH is required to prevent supraphysiological activation of retinoic signaling through maintenance and reinforcement of expression of the Cyp26 genes. Furthermore, in other tissues and organs, disruptions of the Hedgehog or the retinoic acid pathways during development generate similar phenotypes. These findings reveal that rigidly calibrated Hedgehog and retinoic acid activities are required for normal organogenesis and tissue patterning.
Keywords: CRE/LoxP; Cyp26 enzymes; congenital anomalies; hedgehog signaling; mouse models; retinoic acid; smoothened; sonic hedgehog.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- McMahon A.P., Ingham P.W., Tabin T.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003;53:1–114. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
