

Biophysical characterization of full-length human phenylalanine hydroxylase provides a deeper understanding of its quaternary structure equilibrium
- PMID: 31076506
- PMCID: PMC6664189
- DOI: 10.1074/jbc.RA119.008294
Biophysical characterization of full-length human phenylalanine hydroxylase provides a deeper understanding of its quaternary structure equilibrium
Abstract
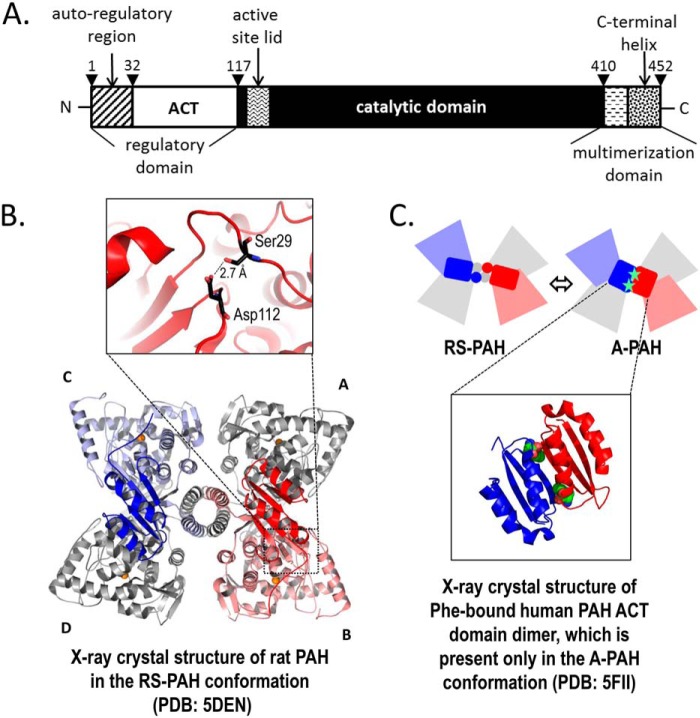
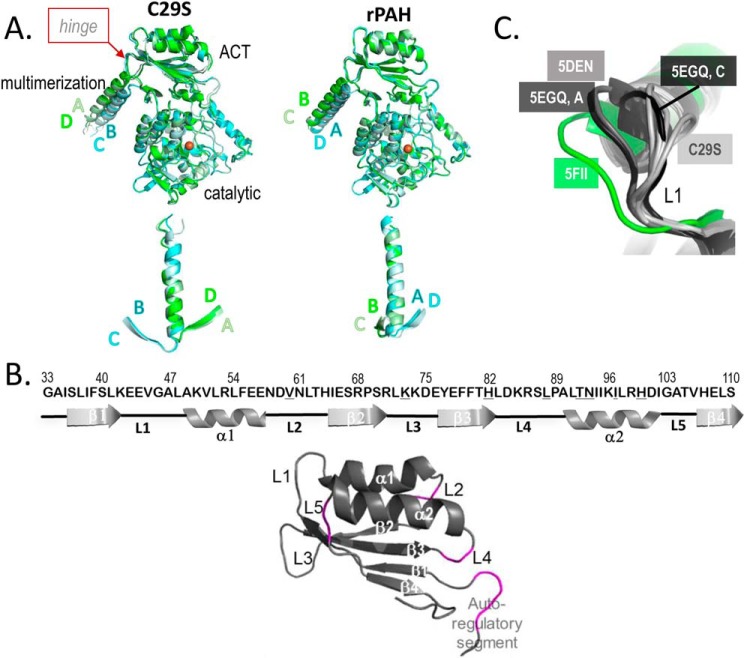
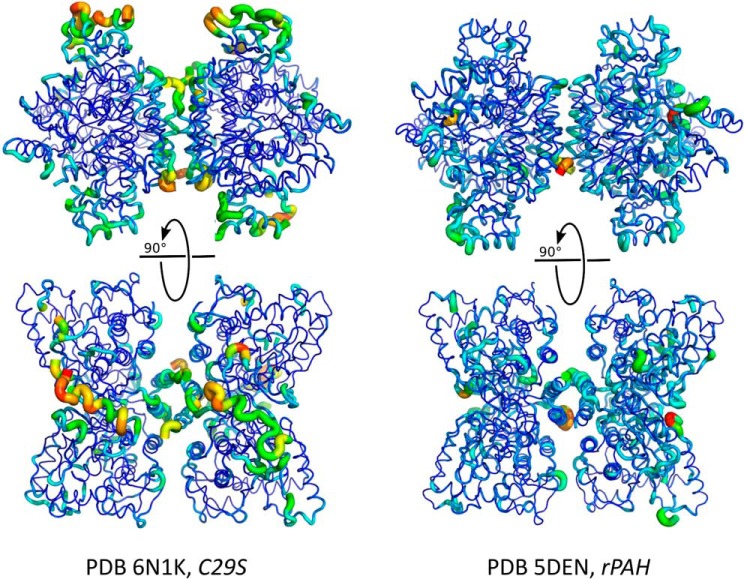
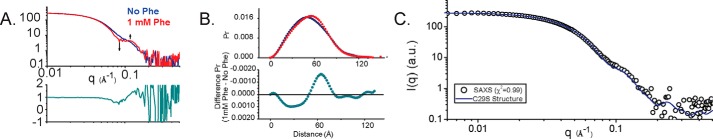
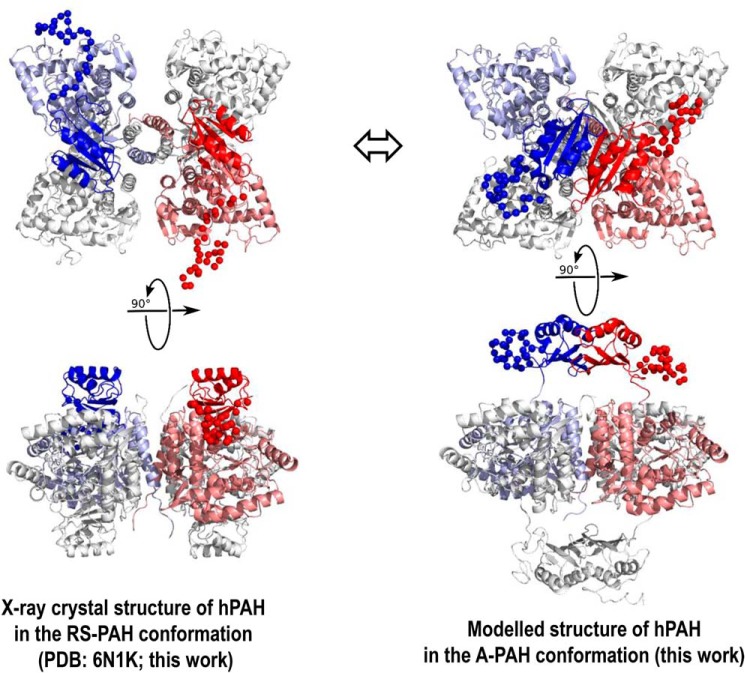
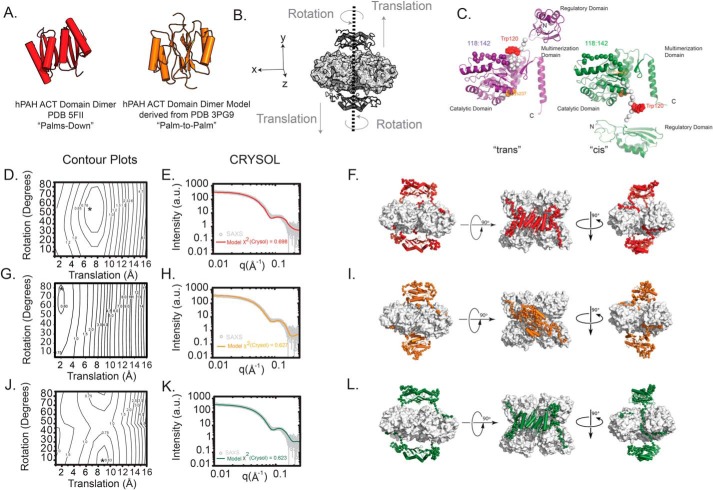
Dysfunction of human phenylalanine hydroxylase (hPAH, EC 1.14.16.1) is the primary cause of phenylketonuria, the most common inborn error of amino acid metabolism. The dynamic domain rearrangements of this multimeric protein have thwarted structural study of the full-length form for decades, until now. In this study, a tractable C29S variant of hPAH (C29S) yielded a 3.06 Å resolution crystal structure of the tetrameric resting-state conformation. We used size-exclusion chromatography in line with small-angle X-ray scattering (SEC-SAXS) to analyze the full-length hPAH solution structure both in the presence and absence of Phe, which serves as both substrate and allosteric activators. Allosteric Phe binding favors accumulation of an activated PAH tetramer conformation, which is biophysically distinct in solution. Protein characterization with enzyme kinetics and intrinsic fluorescence revealed that the C29S variant and hPAH are otherwise equivalent in their response to Phe, further supported by their behavior on various chromatography resins and by analytical ultracentrifugation. Modeling of resting-state and activated forms of C29S against SAXS data with available structural data created and evaluated several new models for the transition between the architecturally distinct conformations of PAH and highlighted unique intra- and inter-subunit interactions. Three best-fitting alternative models all placed the allosteric Phe-binding module 8-10 Å farther from the tetramer center than do all previous models. The structural insights into allosteric activation of hPAH reported here may help inform ongoing efforts to treat phenylketonuria with novel therapeutic approaches.
Keywords: ACT domain; X-ray crystallography; allosteric regulation; inborn error of metabolism; phenylalanine hydroxylase; phenylketonuria; protein conformation; resting-state structure; small-angle X-ray scattering (SAXS); structural biology; structural modeling.
© 2019 Arturo et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

References
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
