

Molecular recording of mammalian embryogenesis
- PMID: 31086336
- PMCID: PMC7229772
- DOI: 10.1038/s41586-019-1184-5
Molecular recording of mammalian embryogenesis
Abstract
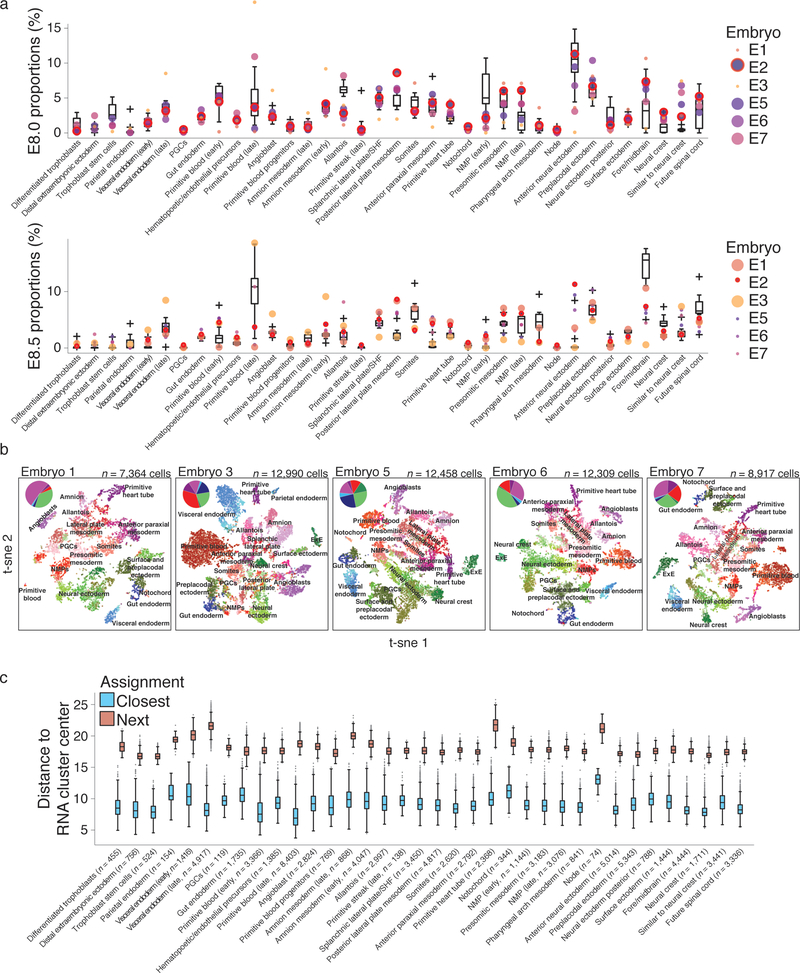
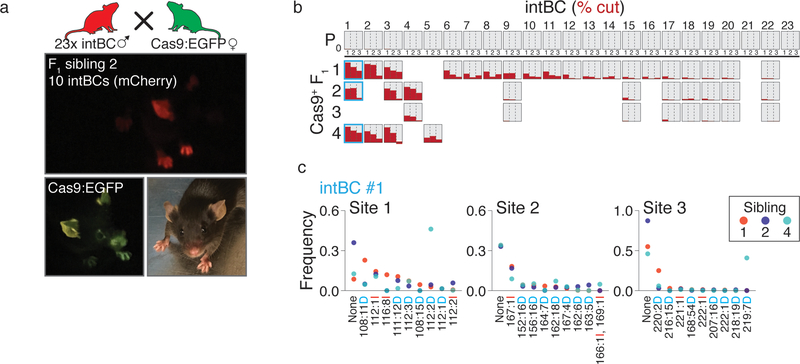
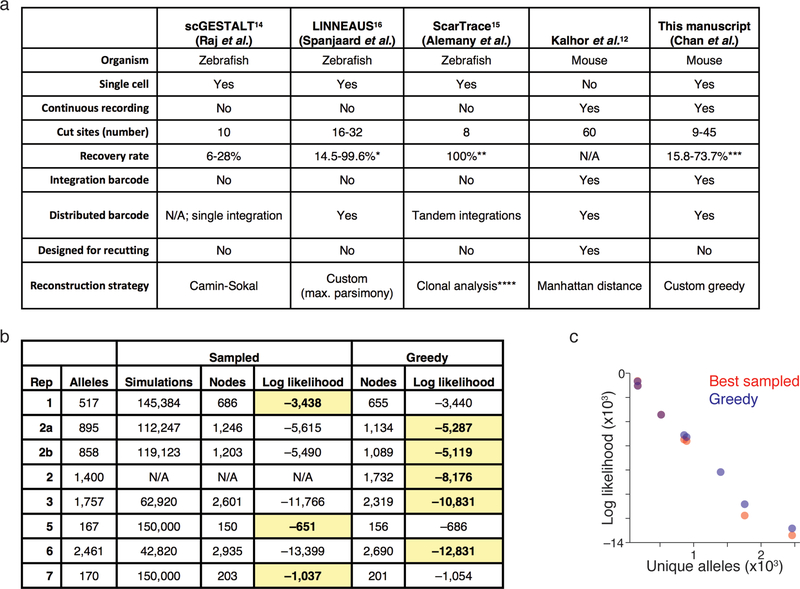
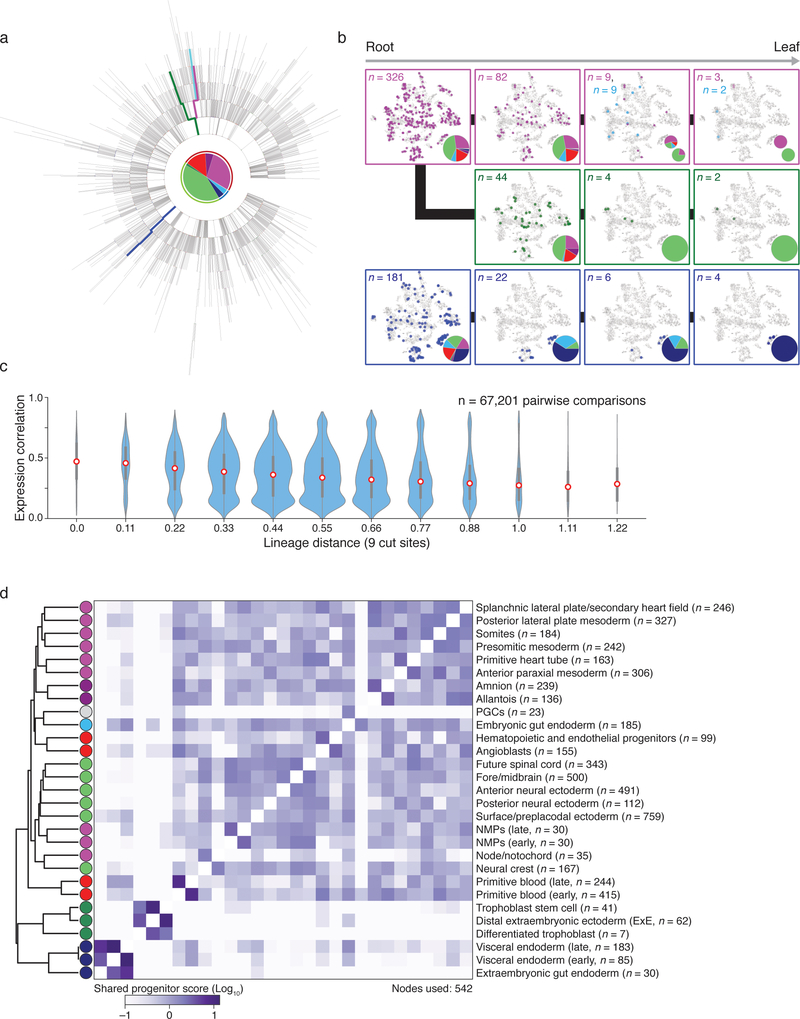
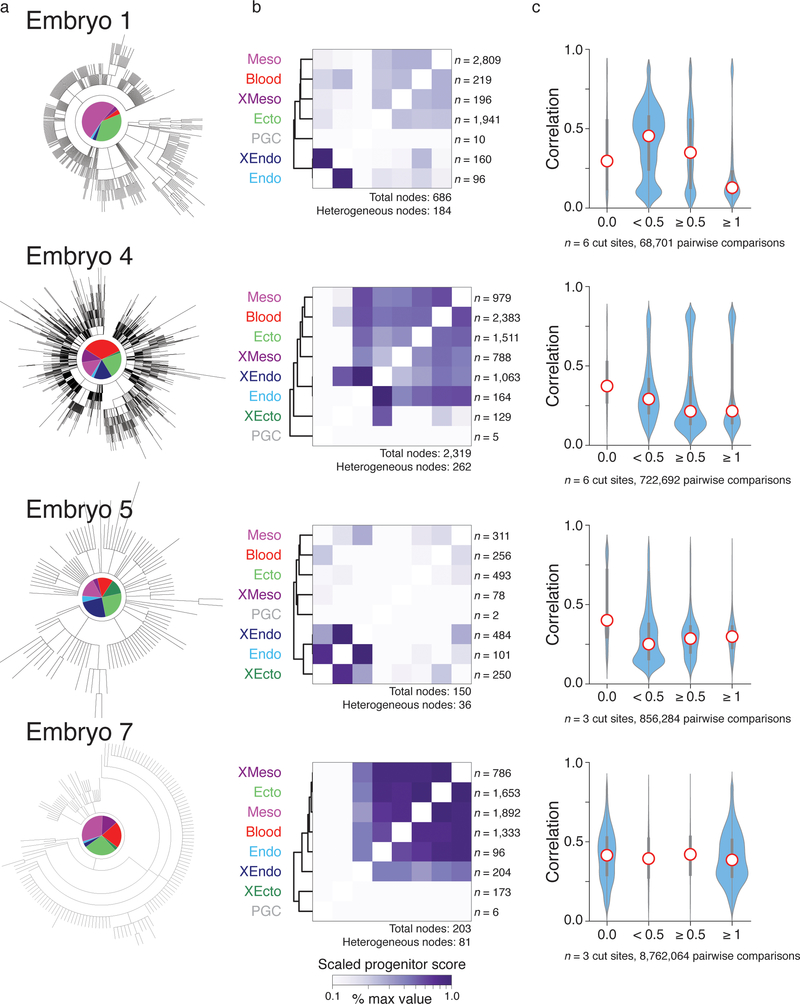
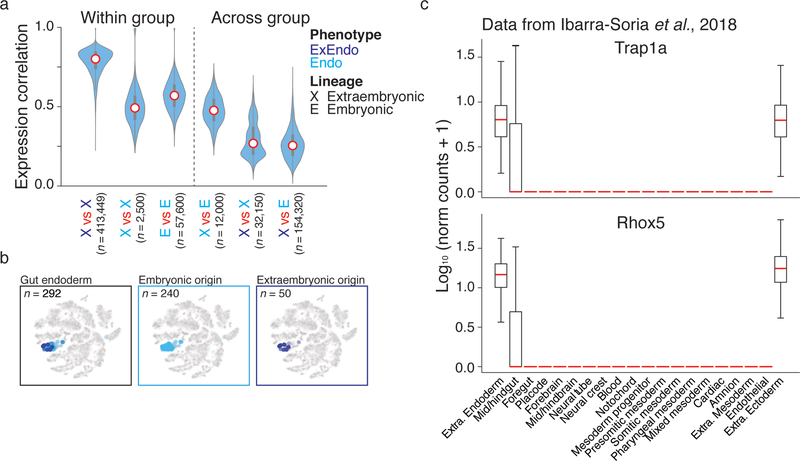
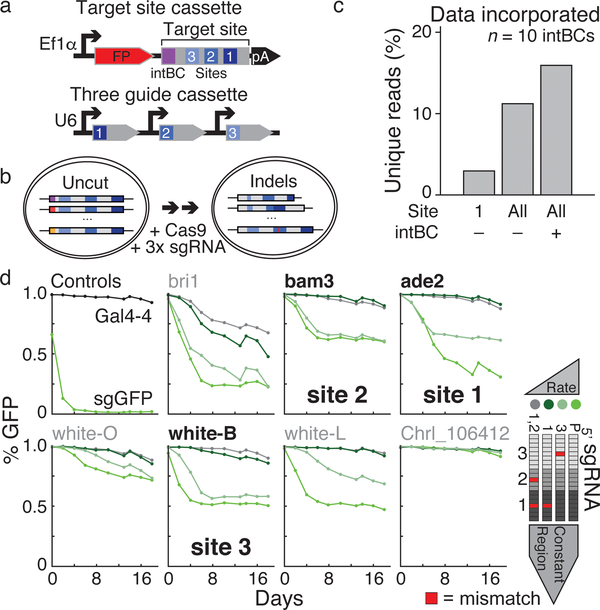
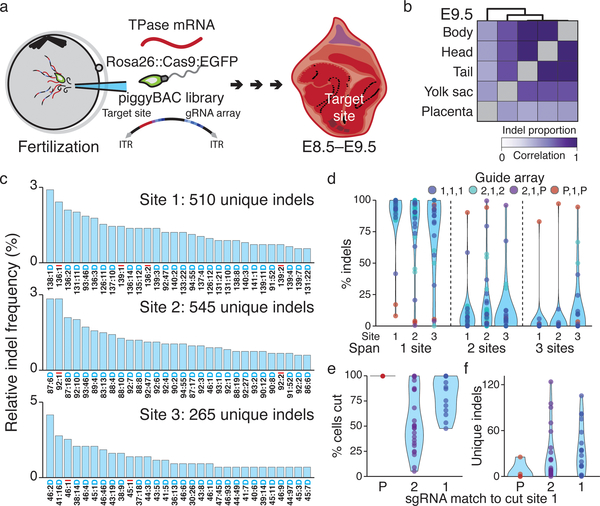
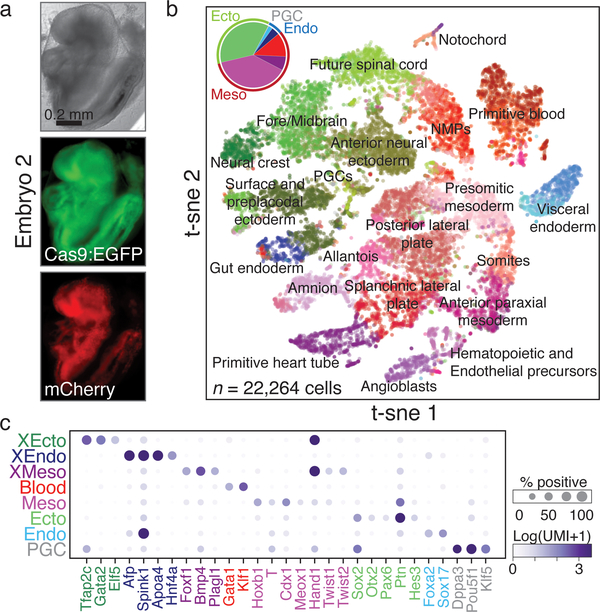
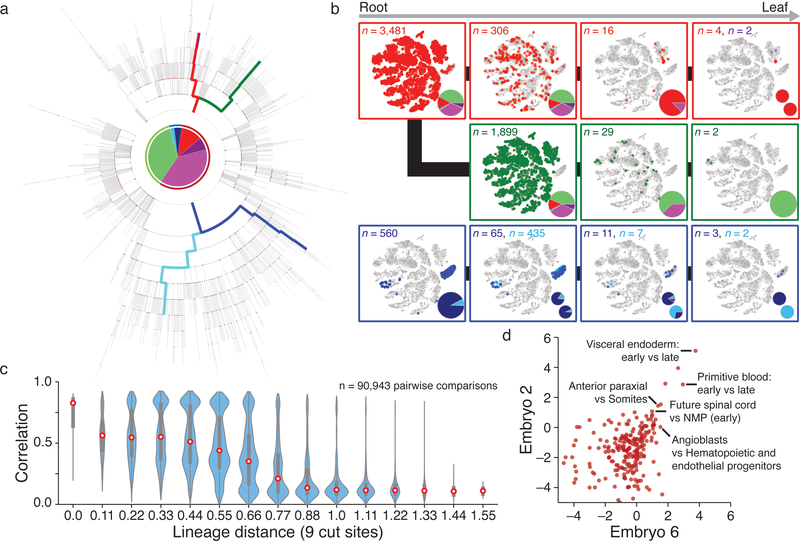
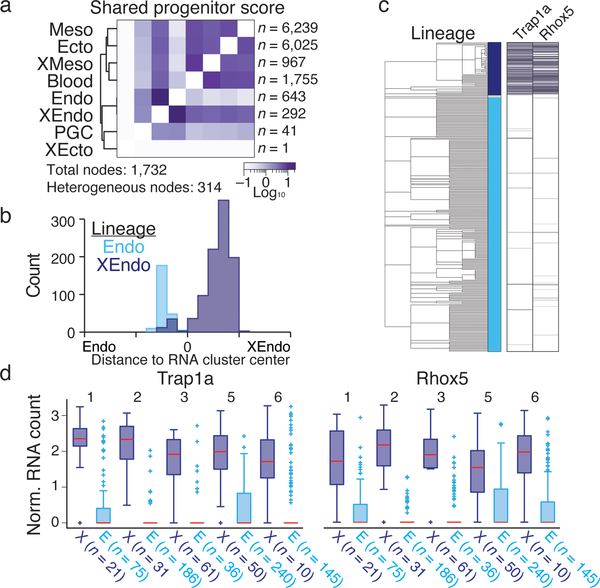
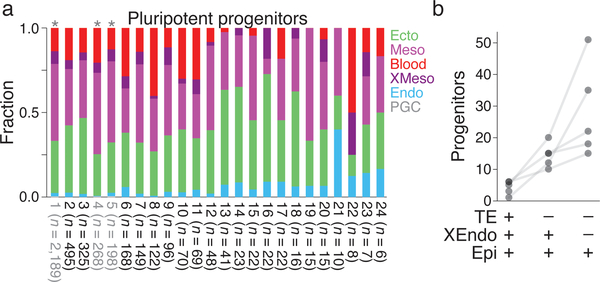
Ontogeny describes the emergence of complex multicellular organisms from single totipotent cells. This field is particularly challenging in mammals, owing to the indeterminate relationship between self-renewal and differentiation, variation in progenitor field sizes, and internal gestation in these animals. Here we present a flexible, high-information, multi-channel molecular recorder with a single-cell readout and apply it as an evolving lineage tracer to assemble mouse cell-fate maps from fertilization through gastrulation. By combining lineage information with single-cell RNA sequencing profiles, we recapitulate canonical developmental relationships between different tissue types and reveal the nearly complete transcriptional convergence of endodermal cells of extra-embryonic and embryonic origins. Finally, we apply our cell-fate maps to estimate the number of embryonic progenitor cells and their degree of asymmetric partitioning during specification. Our approach enables massively parallel, high-resolution recording of lineage and other information in mammalian systems, which will facilitate the construction of a quantitative framework for understanding developmental processes.
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Recording embryogenesis.Nat Rev Genet. 2019 Jul;20(7):373. doi: 10.1038/s41576-019-0141-3. Nat Rev Genet. 2019. PMID: 31114033 No abstract available.
-
Tracing cell fate.Nat Rev Mol Cell Biol. 2019 Aug;20(8):454-455. doi: 10.1038/s41580-019-0148-9. Nat Rev Mol Cell Biol. 2019. PMID: 31114039 No abstract available.
References
-
- Sulston JE, Schierenberg E, White JG & Thomson JN The embryonic cell lineage of the nematode Caenorhabditis elegans. Developmental Biology 100, 64–119 (1983). - PubMed
-
- Pijuan-Sala B, Guibentif C. & Göttgens B. Single-cell transcriptional profiling: a window into embryonic cell-type specification. Nat. Rev. Mol. Cell Biol 19, 399–412 (2018). - PubMed
-
- Zernicka-Goetz M. Patterning of the embryo: the first spatial decisions in the life of a mouse. Development 129, 815–829 (2002). - PubMed
-
- Plass M. et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 360, eaaq1723 (2018). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
