

Genetic Disorders of Manganese Metabolism
- PMID: 31089831
- PMCID: PMC6517356
- DOI: 10.1007/s11910-019-0942-y
Genetic Disorders of Manganese Metabolism
Abstract
Purpose of review: This article provides an overview of the pathogenesis, clinical presentation and treatment of inherited manganese transporter defects.
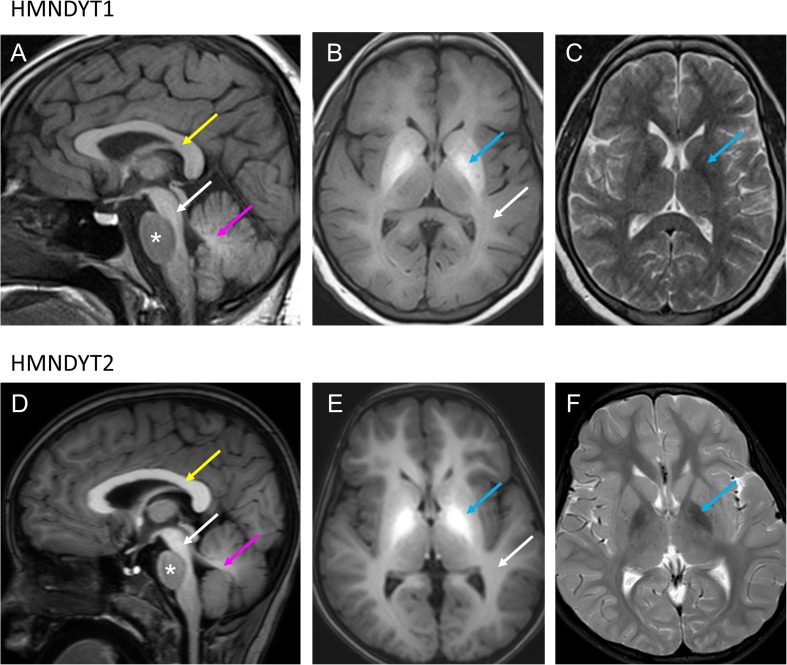
Recent findings: Identification of a new group of manganese transportopathies has greatly advanced our understanding of how manganese homeostasis is regulated in vivo. While the manganese efflux transporter SLC30A10 and the uptake transporter SLC39A14 work synergistically to reduce the manganese load, SLC39A8 has an opposing function facilitating manganese uptake into the organism. Bi-allelic mutations in any of these transporter proteins disrupt the manganese equilibrium and lead to neurological disease: Hypermanganesaemia with dystonia 1 (SLC30A10 deficiency) and hypermanganesaemia with dystonia 2 (SLC39A14 deficiency) are characterised by manganese neurotoxicity while SLC39A8 mutations cause a congenital disorder of glycosylation type IIn due to Mn deficiency. Inherited manganese transporter defects are an important differential diagnosis of paediatric movement disorders. Manganese blood levels and MRI brain are diagnostic and allow early diagnosis to avoid treatment delay.
Keywords: HMNDYT1; HMNDYT2; Manganese; SLC30A10; SLC39A14; SLC39A8.
Conflict of interest statement
The authors declare that they have no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
