

Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in Leishmania
- PMID: 31091230
- PMCID: PMC6519804
- DOI: 10.1371/journal.pgen.1008042
Whole genome sequencing of experimental hybrids supports meiosis-like sexual recombination in Leishmania
Abstract
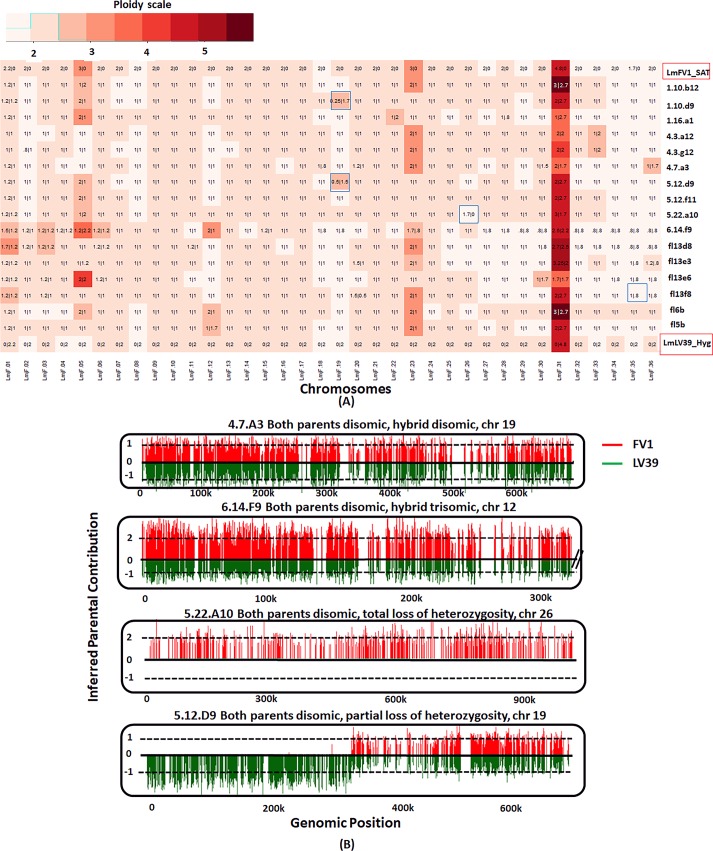
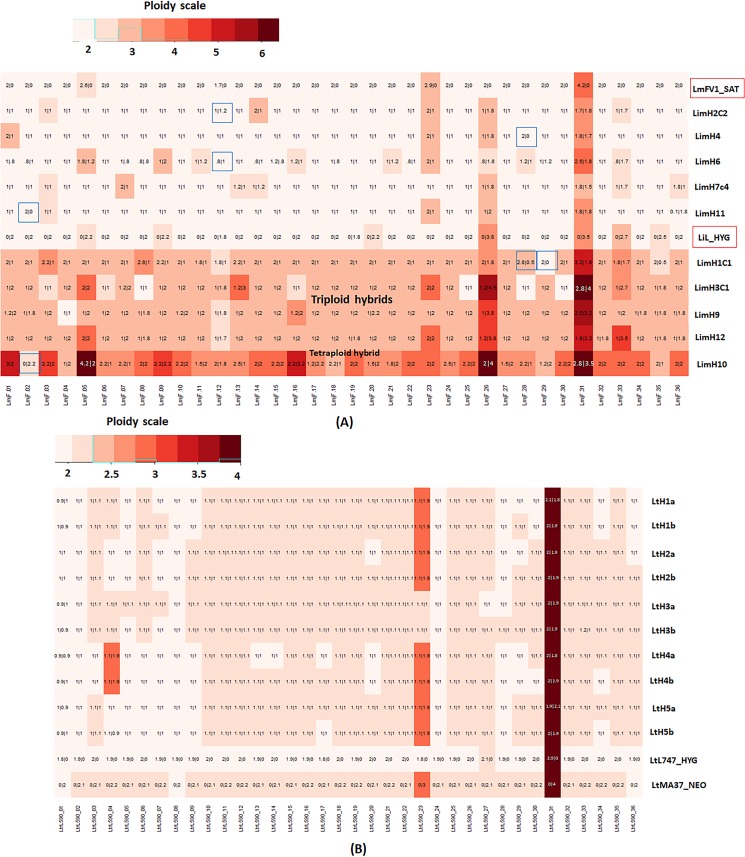
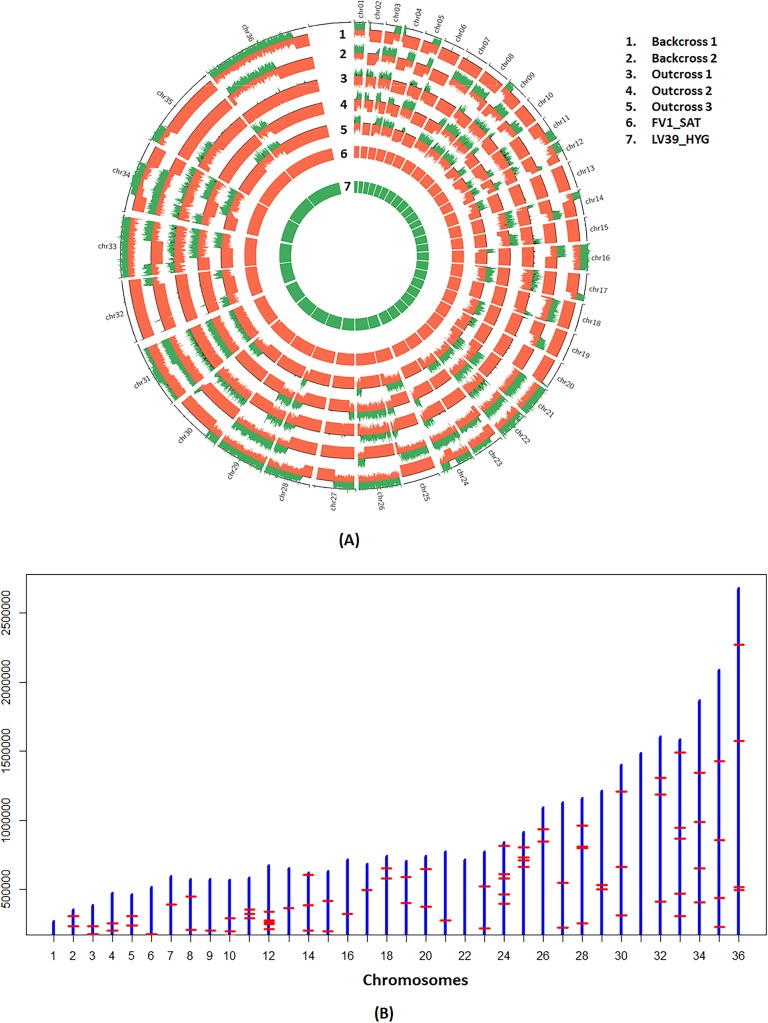
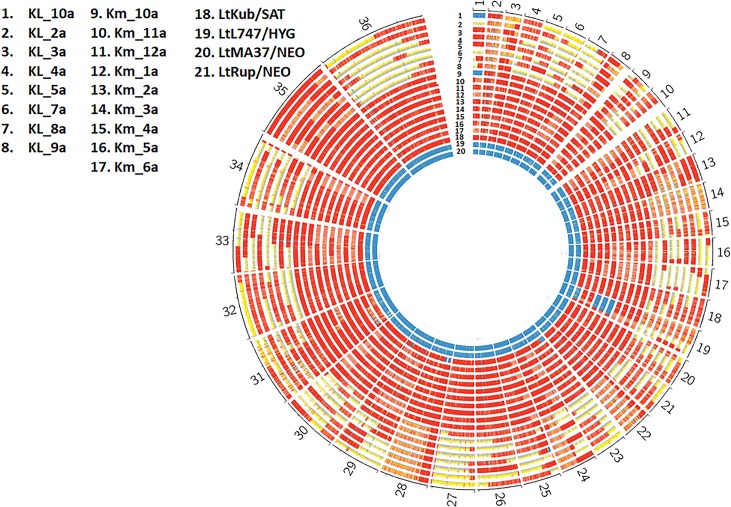
Hybrid genotypes have been repeatedly described among natural isolates of Leishmania, and the recovery of experimental hybrids from sand flies co-infected with different strains or species of Leishmania has formally demonstrated that members of the genus possess the machinery for genetic exchange. As neither gamete stages nor cell fusion events have been directly observed during parasite development in the vector, we have relied on a classical genetic analysis to determine if Leishmania has a true sexual cycle. Here, we used whole genome sequencing to follow the chromosomal inheritance patterns of experimental hybrids generated within and between different strains of L. major and L. infantum. We also generated and sequenced the first experimental hybrids in L. tropica. We found that in each case the parental somy and allele contributions matched the inheritance patterns expected under meiosis 97-99% of the time. The hybrids were equivalent to F1 progeny, heterozygous throughout most of the genome for the markers that were homozygous and different between the parents. Rare, non-Mendelian patterns of chromosomal inheritance were observed, including a gain or loss of somy, and loss of heterozygosity, that likely arose during meiosis or during mitotic divisions of the progeny clones in the fly or culture. While the interspecies hybrids appeared to be sterile, the intraspecies hybrids were able to produce backcross and outcross progeny. Analysis of 5 backcross and outcross progeny clones generated from an L. major F1 hybrid, as well as 17 progeny clones generated from backcrosses involving a natural hybrid of L. tropica, revealed genome wide patterns of recombination, demonstrating that classical crossing over occurs at meiosis, and allowed us to construct the first physical and genetic maps in Leishmania. Altogether, the findings provide strong evidence for meiosis-like sexual recombination in Leishmania, presenting clear opportunities for forward genetic analysis and positional cloning of important genes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
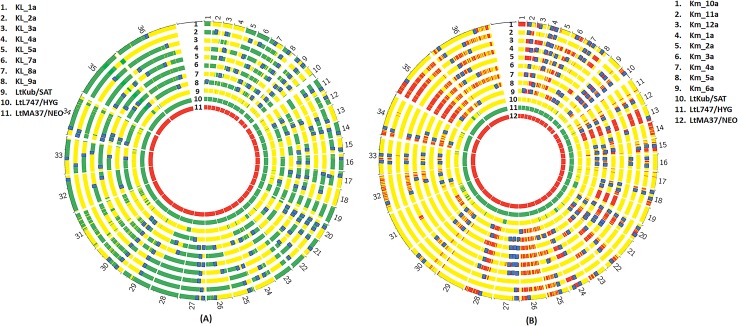
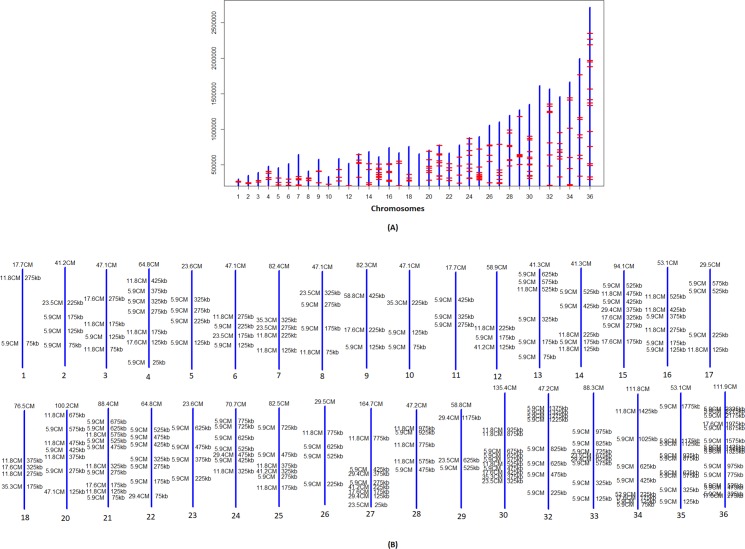
References
-
- Banuls AL, Guerrini F, Le Pont F, Barrera C, Espinel I, Guderian R, et al. Evidence for hybridization by multilocus enzyme electrophoresis and random amplified polymorphic DNA between Leishmania braziliensis and Leishmania panamensis/guyanensis in Ecuador. J Eukaryot Microbiol. 1997;44(5):408–11. Epub 1997/09/26. . - PubMed
-
- Belli AA, Miles MA, Kelly JM. A putative Leishmania panamensis/Leishmania braziliensis hybrid is a causative agent of human cutaneous leishmaniasis in Nicaragua. Parasitology. 1994;109 (Pt 4):435–42. . - PubMed
-
- Dujardin JC, Banuls AL, Llanos-Cuentas A, Alvarez E, DeDoncker S, Jacquet D, et al. Putative Leishmania hybrids in the Eastern Andean valley of Huanuco, Peru. Acta Trop. 1995;59(4):293–307. . - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
