

Growth Factors as Immunotherapeutic Targets in Cardiovascular Disease
- PMID: 31092009
- PMCID: PMC6613384
- DOI: 10.1161/ATVBAHA.119.311994
Growth Factors as Immunotherapeutic Targets in Cardiovascular Disease
Abstract
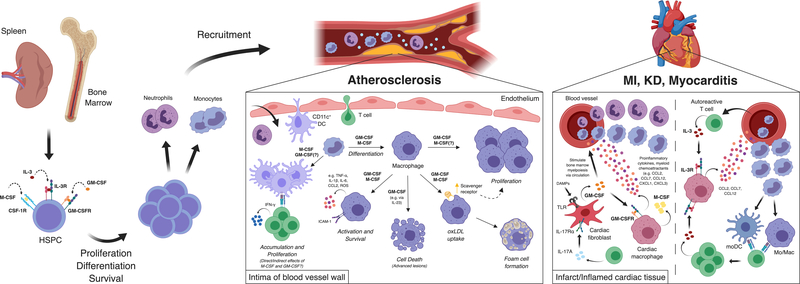
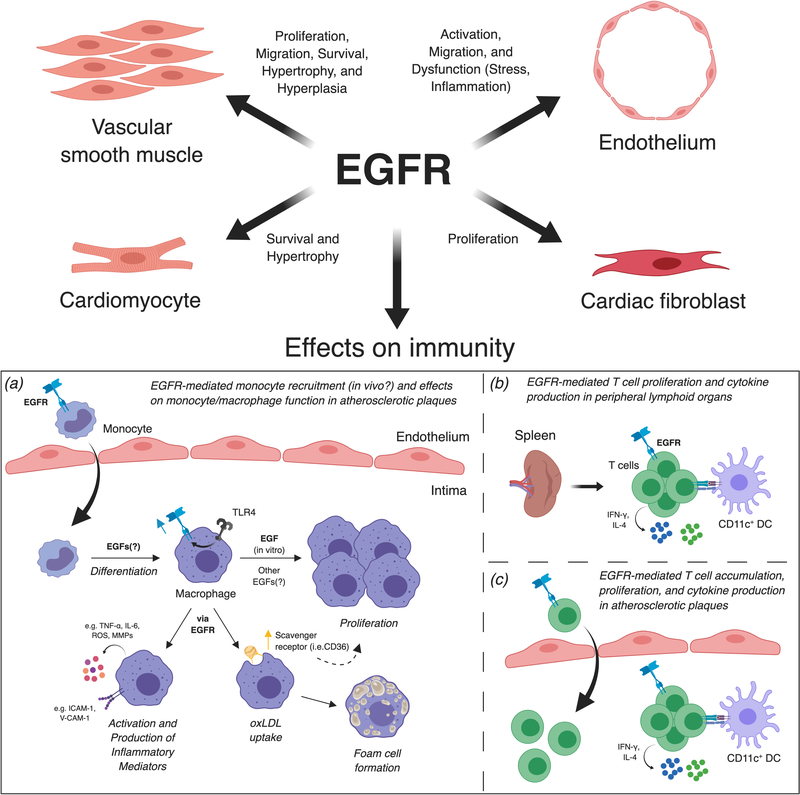
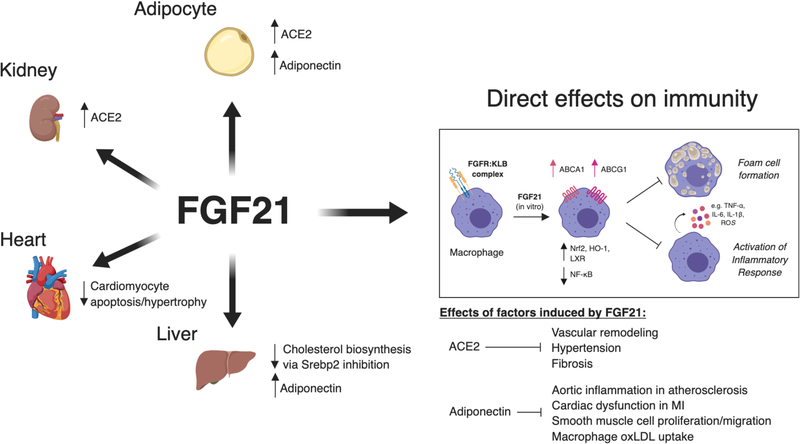
Growth factors, such as CSFs (colony-stimulating factors), EGFs (epidermal growth factors), and FGFs (fibroblast growth factors), are signaling proteins that control a wide range of cellular functions. Although growth factor networks are critical for intercellular communication and tissue homeostasis, their abnormal production or regulation occurs in various pathologies. Clinical strategies that target growth factors or their receptors are used to treat a variety of conditions but have yet to be adopted for cardiovascular disease. In this review, we focus on M-CSF (macrophage-CSF), GM-CSF (granulocyte-M-CSF), IL (interleukin)-3, EGFR (epidermal growth factor receptor), and FGF21 (fibroblast growth factor 21). We first discuss the efficacy of targeting these growth factors in other disease contexts (ie, inflammatory/autoimmune diseases, cancer, or metabolic disorders) and then consider arguments for or against targeting them to treat cardiovascular disease. Visual Overview- An online visual overview is available for this article.
Keywords: atherosclerosis; immunotherapy; inflammation; myeloid cells.
Figures
Similar articles
-
Regulation of myeloid development and function by colony stimulating factors.Dev Comp Immunol. 2004 May 3;28(5):509-54. doi: 10.1016/j.dci.2003.09.010. Dev Comp Immunol. 2004. PMID: 15062647 Review.
-
Granulocyte-colony stimulating factor, granulocyte-macrophage colony stimulating factor, PIXY-321, stem cell factor, interleukin-3, and interleukin-7: receptor binding and effects on clonogenic proliferation in acute lymphoblastic leukemia.Leuk Lymphoma. 1994 Dec;16(1-2):79-88. doi: 10.3109/10428199409114143. Leuk Lymphoma. 1994. PMID: 7535143
-
Activities of granulocyte-macrophage colony-stimulating factor and interleukin-3 on monocytes.Am J Hematol. 2004 Apr;75(4):179-89. doi: 10.1002/ajh.20010. Am J Hematol. 2004. PMID: 15054806
-
Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation.J Immunol. 2007 Apr 15;178(8):5245-52. doi: 10.4049/jimmunol.178.8.5245. J Immunol. 2007. PMID: 17404308
-
Clinical toxicity of cytokines used as haemopoietic growth factors.Drug Saf. 1995 Dec;13(6):371-406. doi: 10.2165/00002018-199513060-00006. Drug Saf. 1995. PMID: 8652081 Review.
Cited by
-
Immunotherapeutic Strategies in Cancer and Atherosclerosis-Two Sides of the Same Coin.Front Cardiovasc Med. 2022 Jan 13;8:812702. doi: 10.3389/fcvm.2021.812702. eCollection 2021. Front Cardiovasc Med. 2022. PMID: 35097027 Free PMC article. Review.
-
Emerging Concepts of Vascular Cell Clonal Expansion in Atherosclerosis.Arterioscler Thromb Vasc Biol. 2022 Mar;42(3):e74-e84. doi: 10.1161/ATVBAHA.121.316093. Epub 2022 Feb 3. Arterioscler Thromb Vasc Biol. 2022. PMID: 35109671 Free PMC article. Review.
-
ADAM10 and ADAM17, Major Regulators of Chronic Kidney Disease Induced Atherosclerosis?Int J Mol Sci. 2023 Apr 15;24(8):7309. doi: 10.3390/ijms24087309. Int J Mol Sci. 2023. PMID: 37108478 Free PMC article. Review.
-
Metabolic syndrome.Nat Rev Dis Primers. 2024 Oct 17;10(1):77. doi: 10.1038/s41572-024-00563-5. Nat Rev Dis Primers. 2024. PMID: 39420195 Review.
-
Hypermethylation Effects of Yiqihuoxue Decoction in Diabetic Atherosclerosis Using Genome-Wide DNA Methylation Analyses.J Inflamm Res. 2022 Jan 8;15:163-176. doi: 10.2147/JIR.S335374. eCollection 2022. J Inflamm Res. 2022. PMID: 35035227 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
