

Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis
- PMID: 31092817
- PMCID: PMC6520353
- DOI: 10.1038/s41467-019-09976-3
Joint sequencing of human and pathogen genomes reveals the genetics of pneumococcal meningitis
Abstract
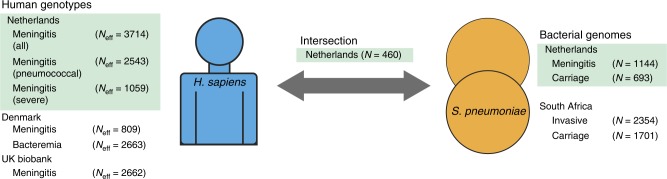
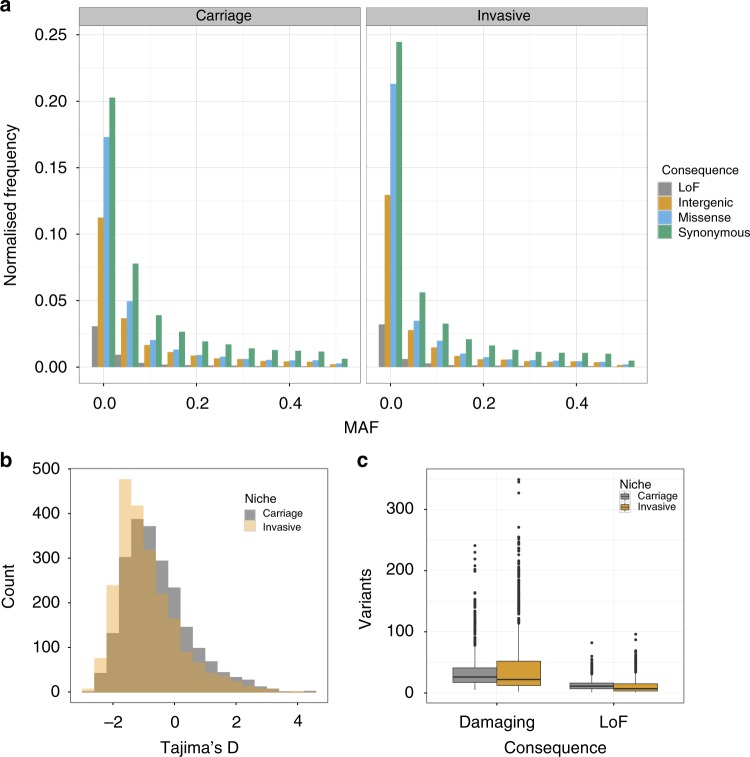
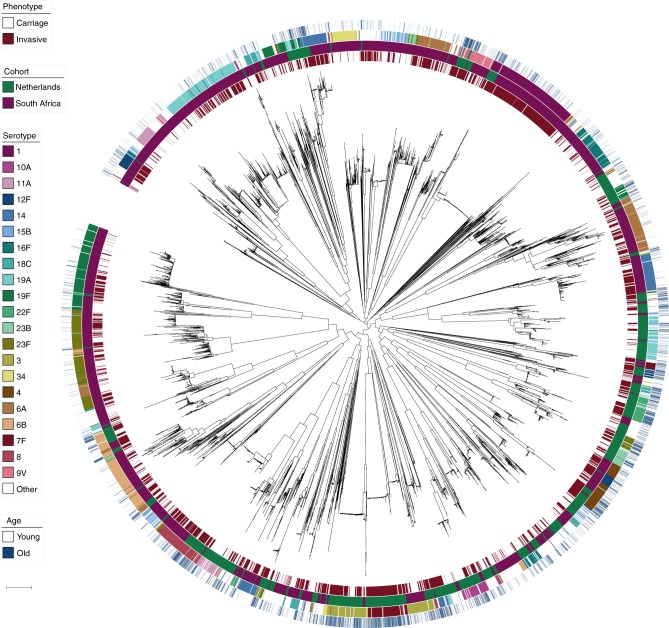
Streptococcus pneumoniae is a common nasopharyngeal colonizer, but can also cause life-threatening invasive diseases such as empyema, bacteremia and meningitis. Genetic variation of host and pathogen is known to play a role in invasive pneumococcal disease, though to what extent is unknown. In a genome-wide association study of human and pathogen we show that human variation explains almost half of variation in susceptibility to pneumococcal meningitis and one-third of variation in severity, identifying variants in CCDC33 associated with susceptibility. Pneumococcal genetic variation explains a large amount of invasive potential (70%), but has no effect on severity. Serotype alone is insufficient to explain invasiveness, suggesting other pneumococcal factors are involved in progression to invasive disease. We identify pneumococcal genes involved in invasiveness including pspC and zmpD, and perform a human-bacteria interaction analysis. These genes are potential candidates for the development of more broadly-acting pneumococcal vaccines.
Conflict of interest statement
N.J.C. and S.D.B. were consultants for Antigen Discovery, Inc involved in the design of a proteome array for
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
