

Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia
- PMID: 31092926
- PMCID: PMC6533116
- DOI: 10.1038/s41586-019-1198-z
Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia
Abstract
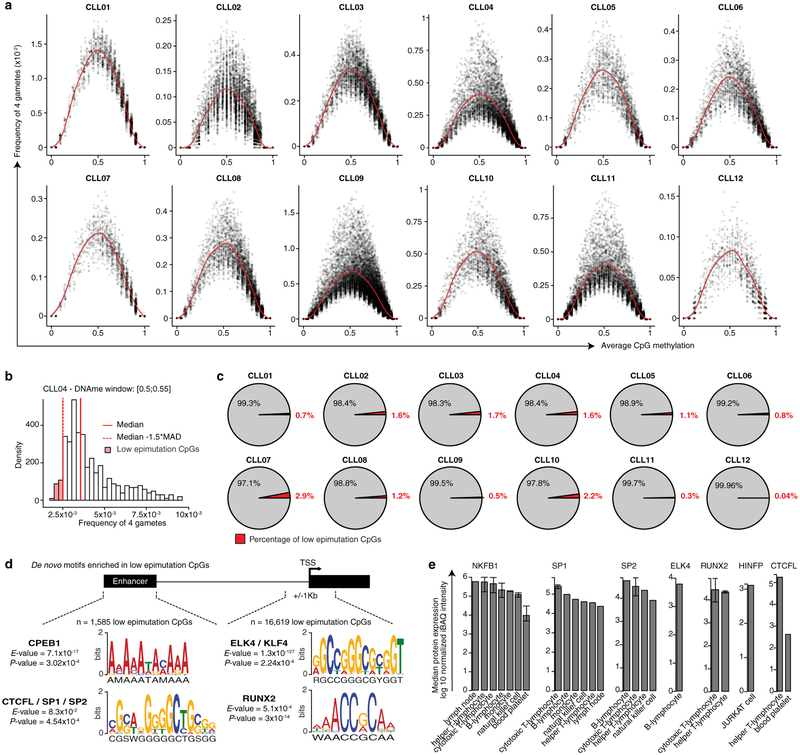
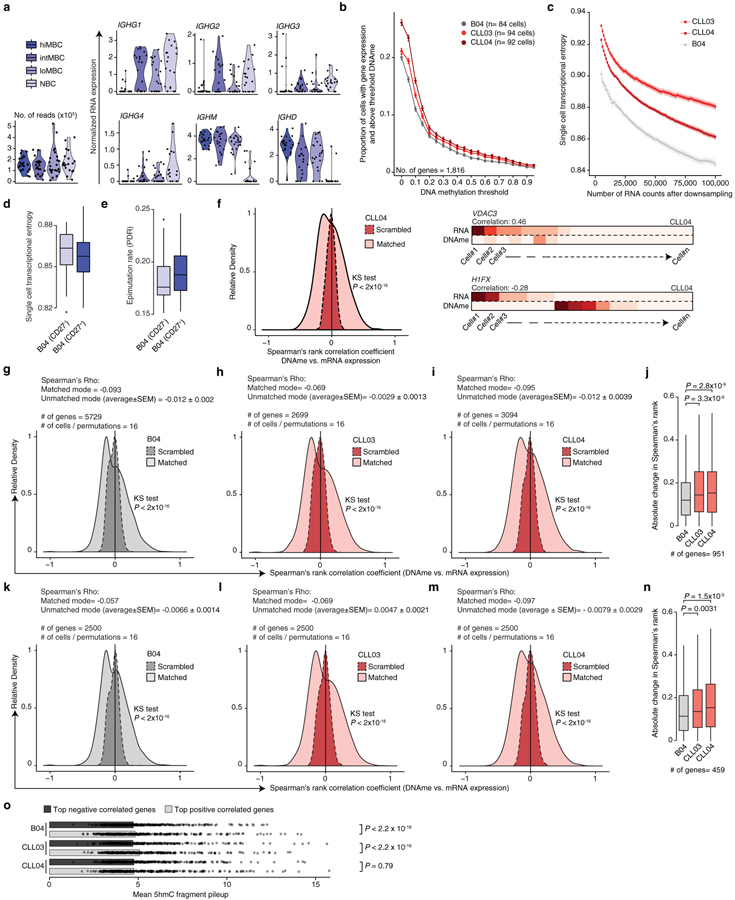
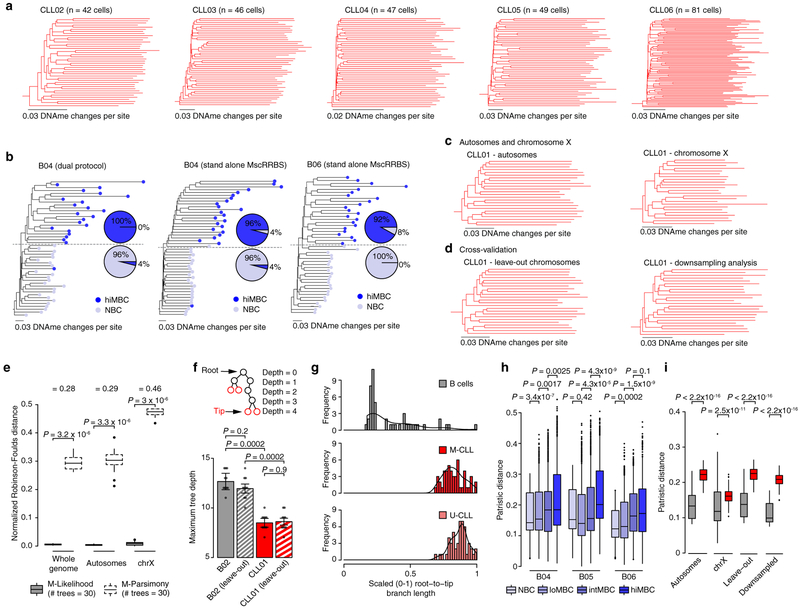
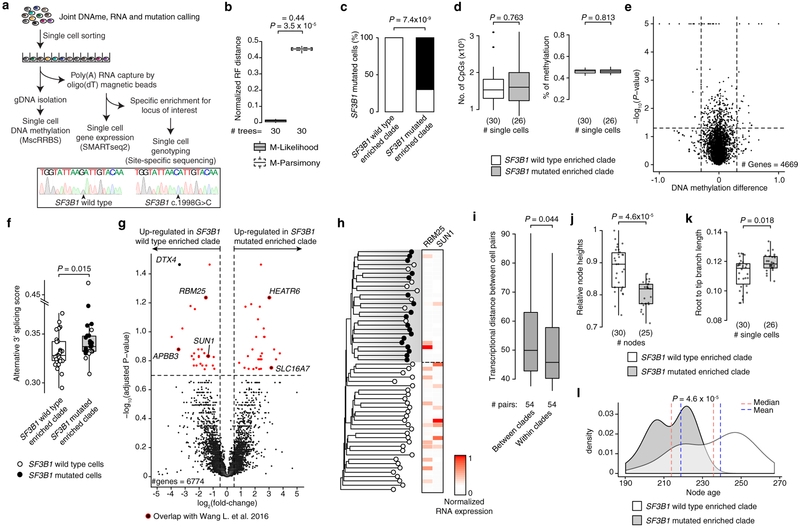
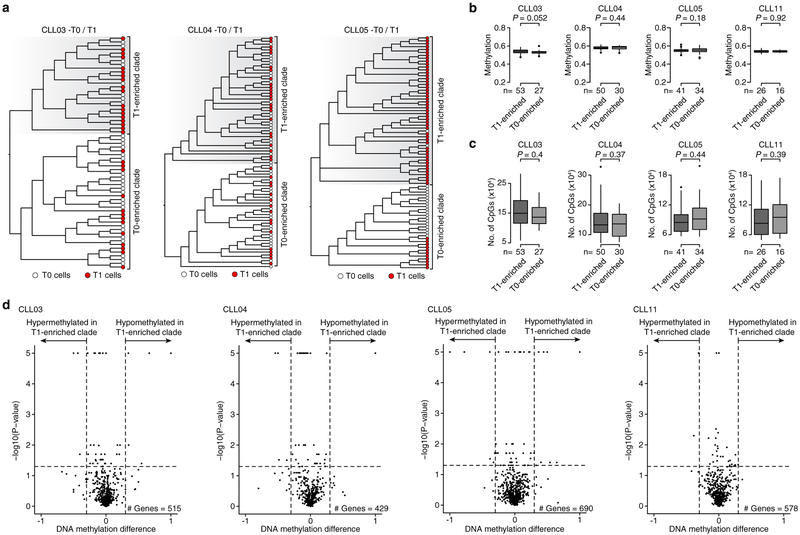
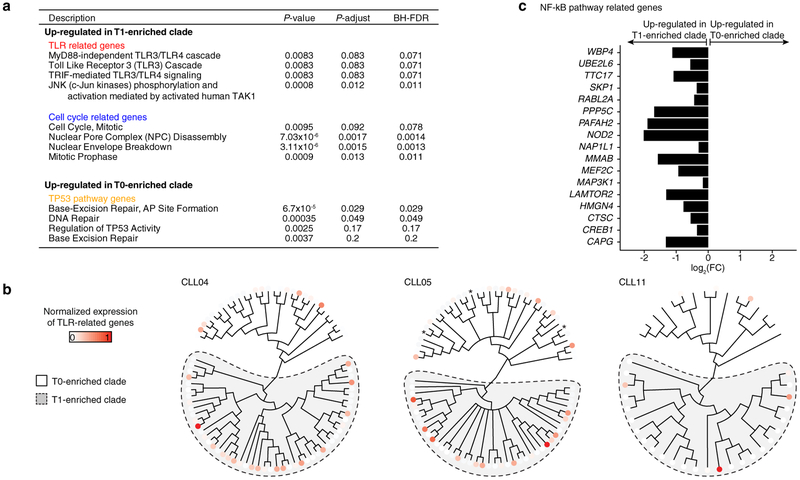
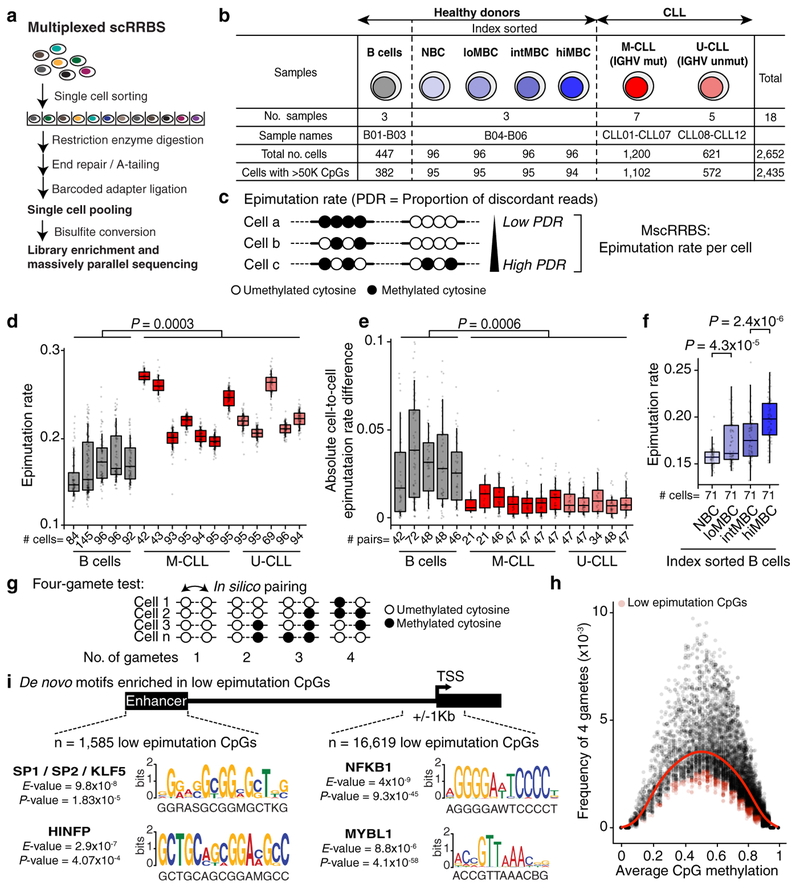
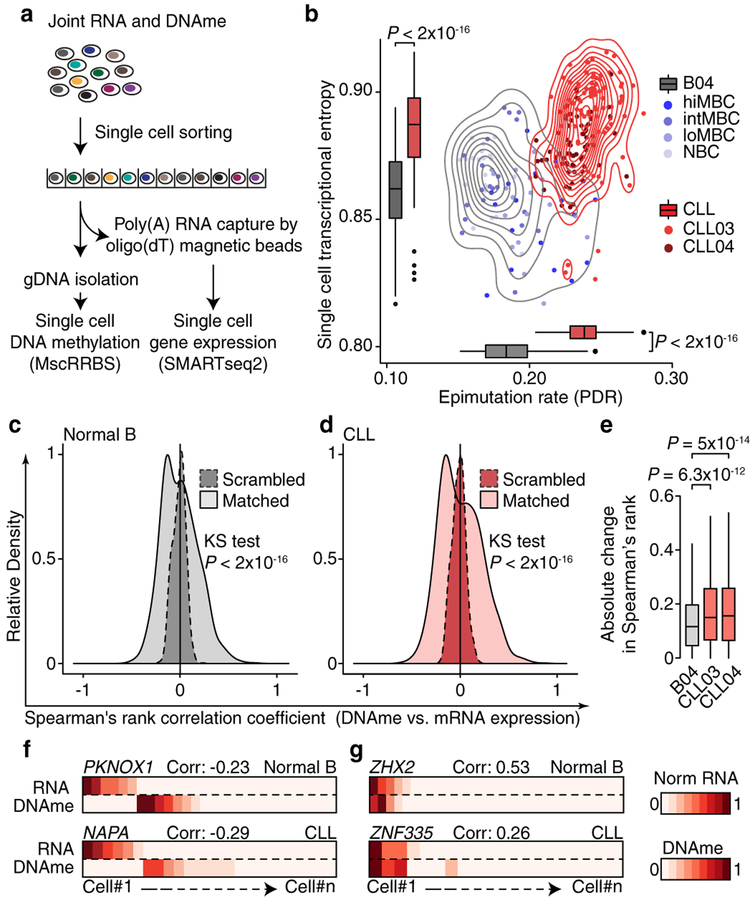
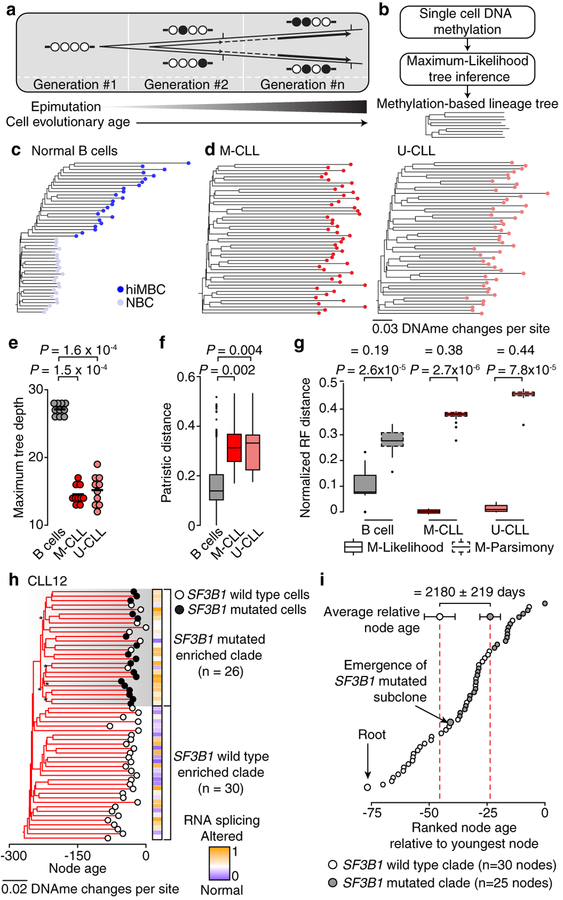
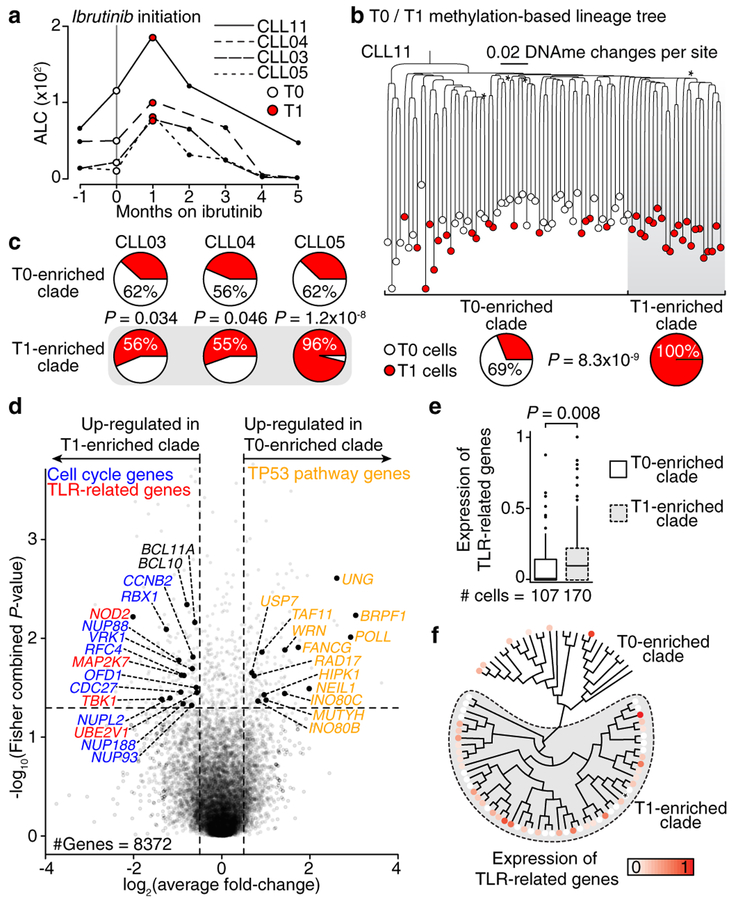
Genetic and epigenetic intra-tumoral heterogeneity cooperate to shape the evolutionary course of cancer1. Chronic lymphocytic leukaemia (CLL) is a highly informative model for cancer evolution as it undergoes substantial genetic diversification and evolution after therapy2,3. The CLL epigenome is also an important disease-defining feature4,5, and growing populations of cells in CLL diversify by stochastic changes in DNA methylation known as epimutations6. However, previous studies using bulk sequencing methods to analyse the patterns of DNA methylation were unable to determine whether epimutations affect CLL populations homogeneously. Here, to measure the epimutation rate at single-cell resolution, we applied multiplexed single-cell reduced-representation bisulfite sequencing to B cells from healthy donors and patients with CLL. We observed that the common clonal origin of CLL results in a consistently increased epimutation rate, with low variability in the cell-to-cell epimutation rate. By contrast, variable epimutation rates across healthy B cells reflect diverse evolutionary ages across the trajectory of B cell differentiation, consistent with epimutations serving as a molecular clock. Heritable epimutation information allowed us to reconstruct lineages at high-resolution with single-cell data, and to apply this directly to patient samples. The CLL lineage tree shape revealed earlier branching and longer branch lengths than in normal B cells, reflecting rapid drift after the initial malignant transformation and a greater proliferative history. Integration of single-cell bisulfite sequencing analysis with single-cell transcriptomes and genotyping confirmed that genetic subclones mapped to distinct clades, as inferred solely on the basis of epimutation information. Finally, to examine potential lineage biases during therapy, we profiled serial samples during ibrutinib-associated lymphocytosis, and identified clades of cells that were preferentially expelled from the lymph node after treatment, marked by distinct transcriptional profiles. The single-cell integration of genetic, epigenetic and transcriptional information thus charts the lineage history of CLL and its evolution with therapy.
Conflict of interest statement
Competing interests
The authors declare no competing financial interests.
Figures
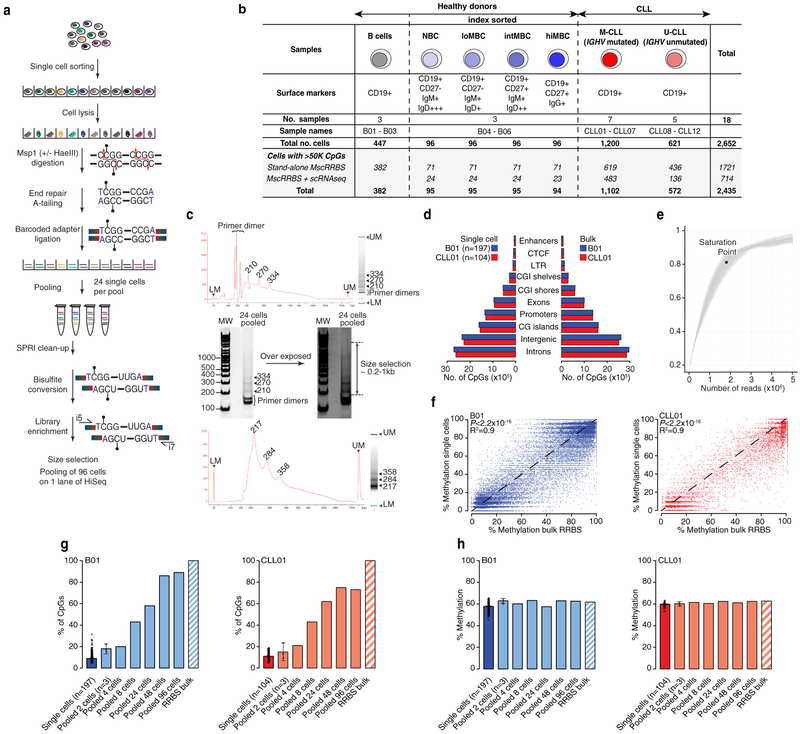
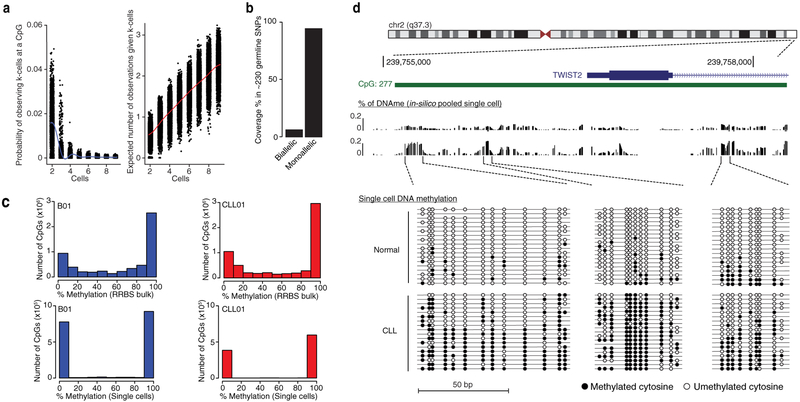
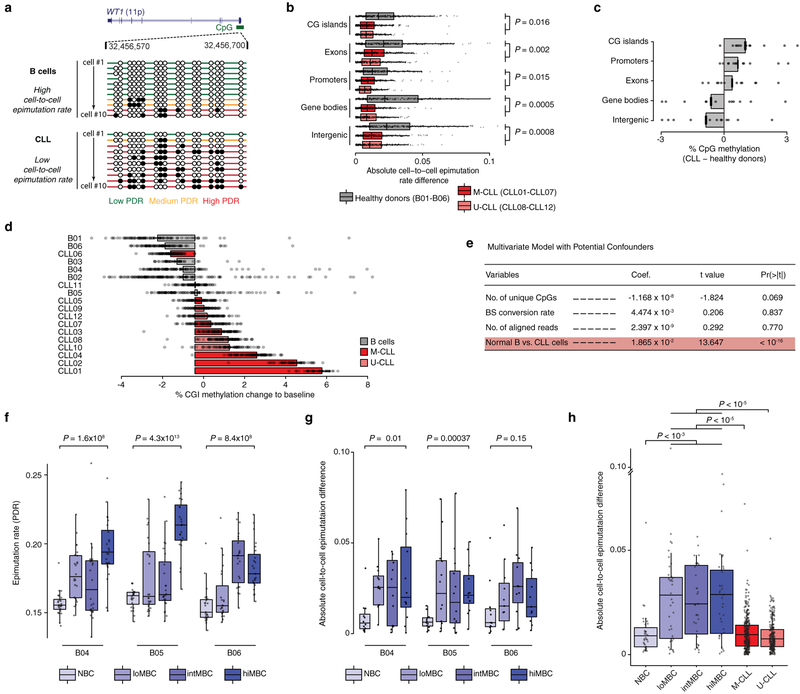
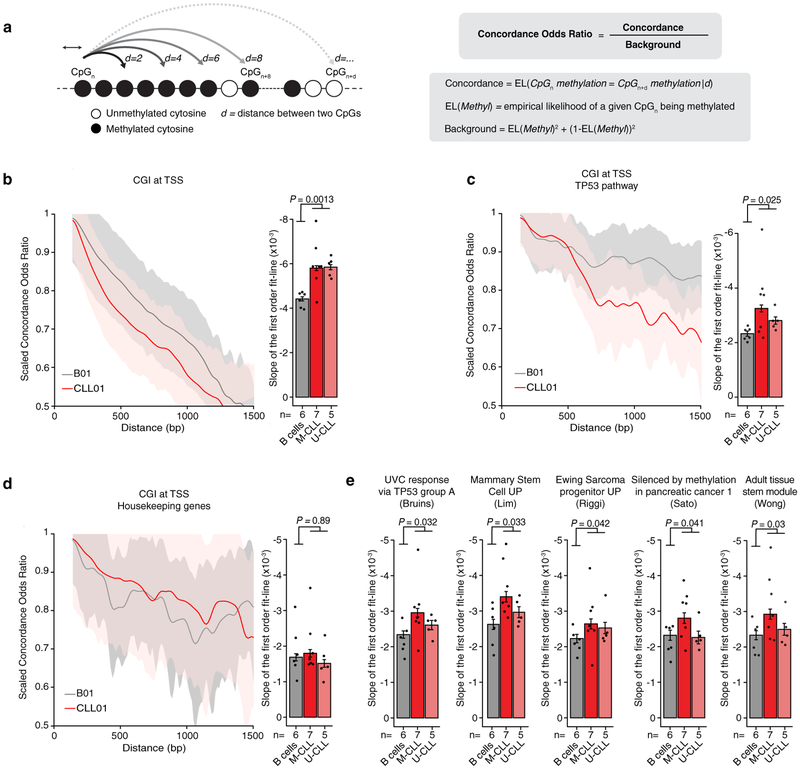
Comment in
-
Epigenetic cancer evolution, one cell at a time.Nat Rev Genet. 2019 Aug;20(8):434-435. doi: 10.1038/s41576-019-0143-1. Nat Rev Genet. 2019. PMID: 31160791 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
