

Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain
- PMID: 31095601
- PMCID: PMC6522025
- DOI: 10.1371/journal.pone.0216539
Whole blood transcriptomic profiles can differentiate vulnerability to chronic low back pain
Abstract
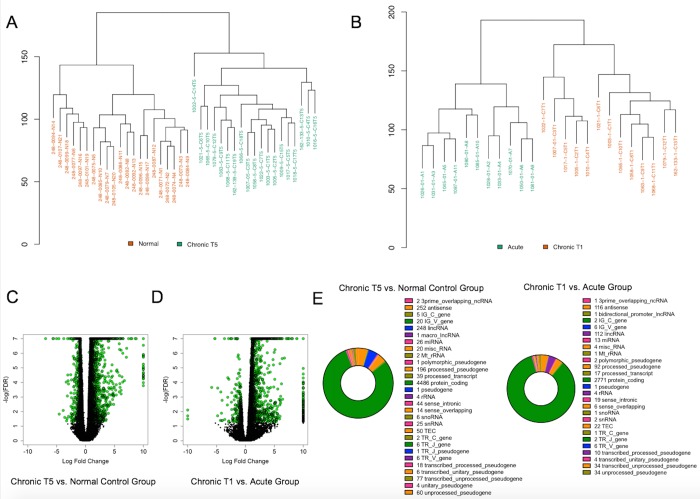
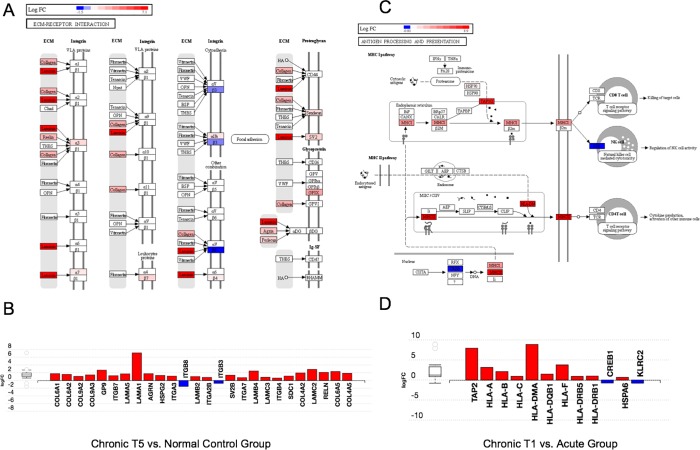
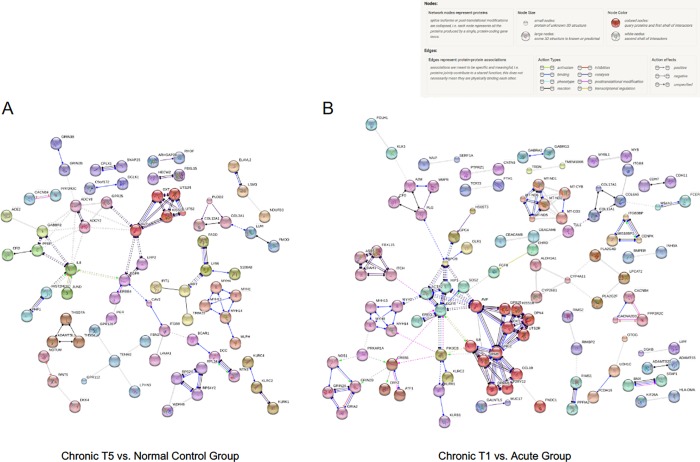
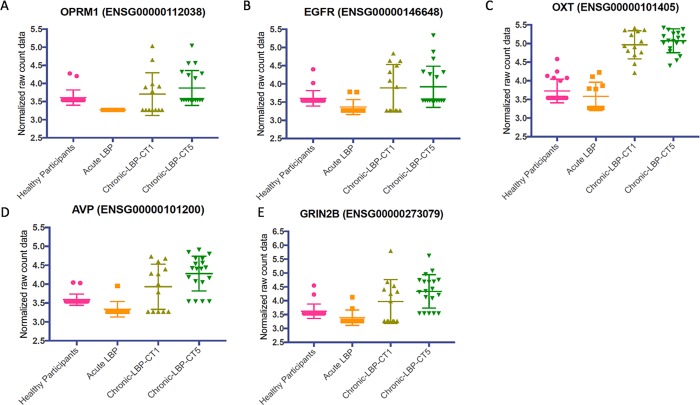
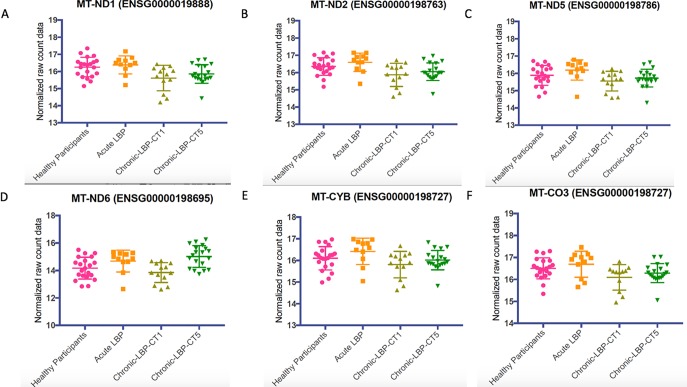
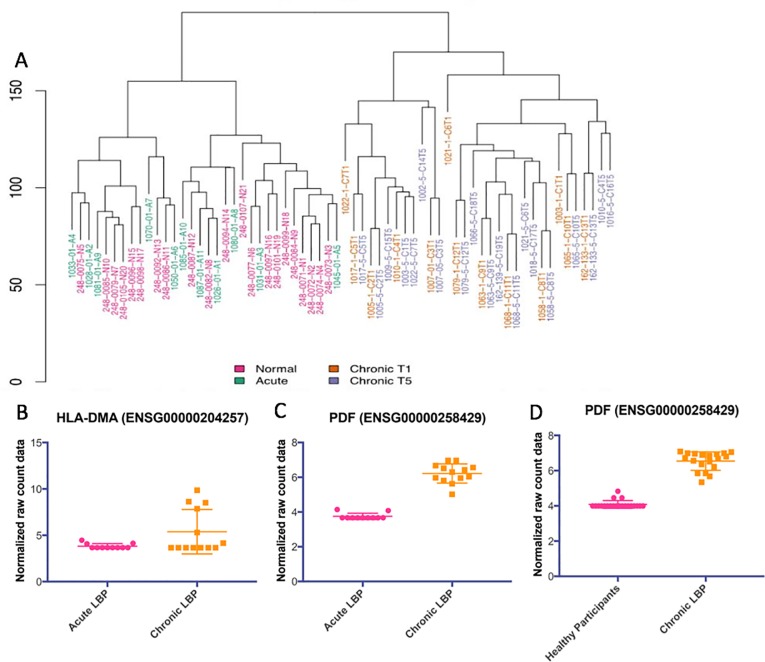
The mechanisms underlying the transition from acute to chronic pain remain unclear. Here, we sought to characterize the transcriptome associated with chronic low back pain as well as the transcriptome of the transition from acute to chronic low back pain. For the analysis, we compared the whole blood transcriptome of: (a) patients at the onset of low back pain who no longer had pain within 6 weeks after onset (acute) with patients who developed chronic low back pain at 6 months (chronic T5); and, (b) patients at the onset of low back pain (chronic T1) who developed chronic pain at 6 months with healthy pain-free (normal) controls. The majority of differentially expressed genes were protein coding. We illustrate a unique chronic low back pain transcriptome characterized by significant enrichment for known pain genes, extracellular matrix genes, and genes from the extended major histocompatibility complex (MHC) genomic locus. The transcriptome of the transition from acute to chronic low back pain was characterized by significant upregulation of antigen presentation pathway (MHC class I and II) genes and downregulation of mitochondrial genes associated with oxidative phosphorylation, suggesting a unique genomic signature of vulnerability to low back pain chronicity.
Conflict of interest statement
SGD, CLR, MG, and ARS have a patent pending (62/607,969 filed 12/20/17) on Biomarkers of Acute and Chronic Pain. The other authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
