

Surgical preparations, labeling strategies, and optical techniques for cell-resolved, in vivo imaging in the mouse spinal cord
- PMID: 31095935
- PMCID: PMC6588420
- DOI: 10.1016/j.expneurol.2019.05.010
Surgical preparations, labeling strategies, and optical techniques for cell-resolved, in vivo imaging in the mouse spinal cord
Abstract
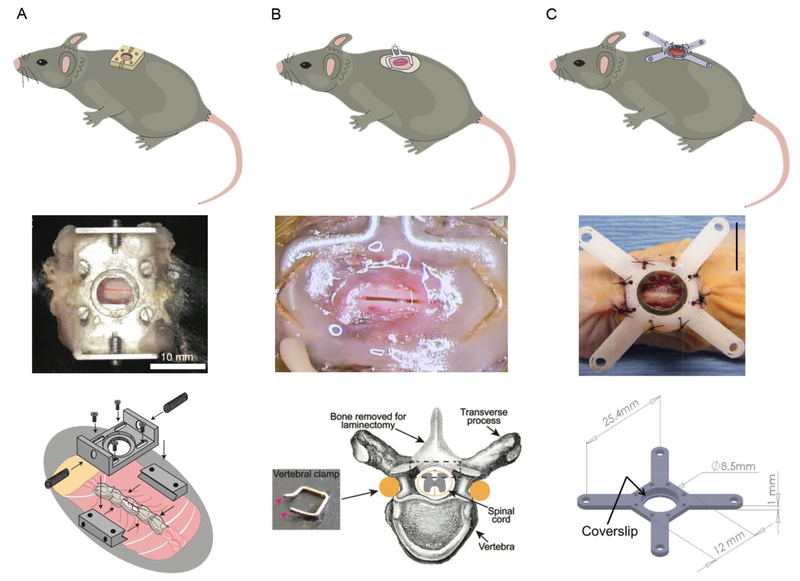
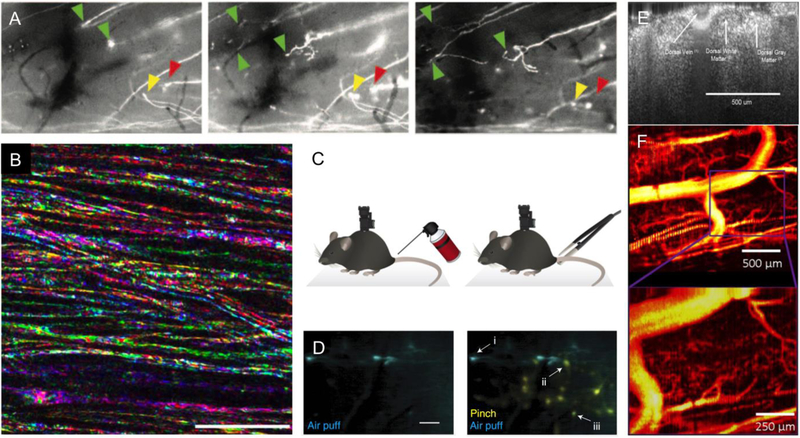
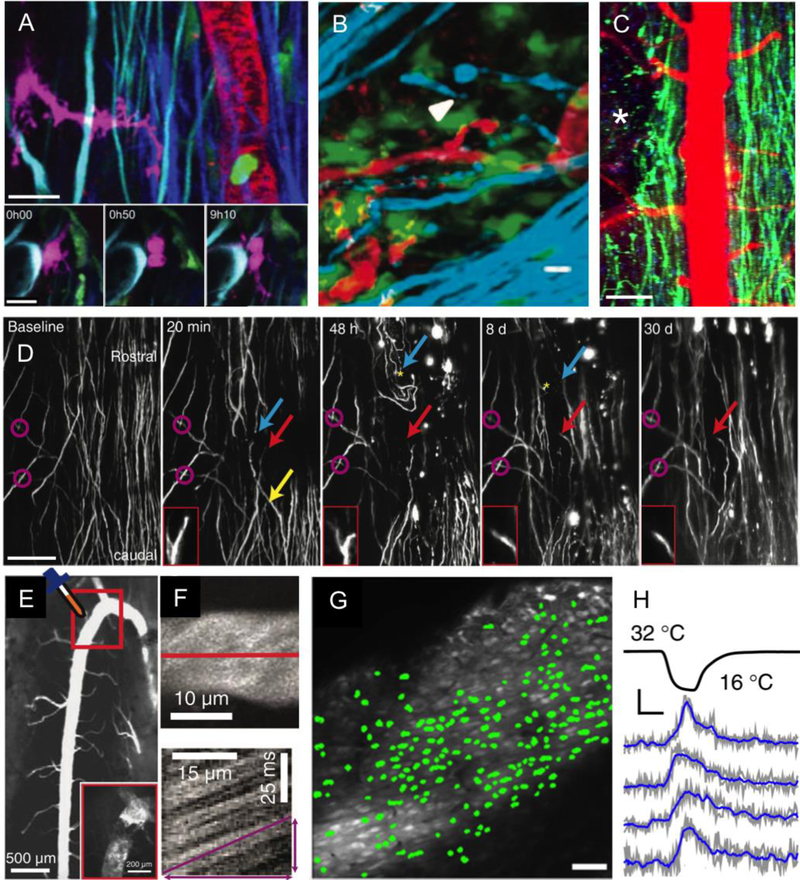
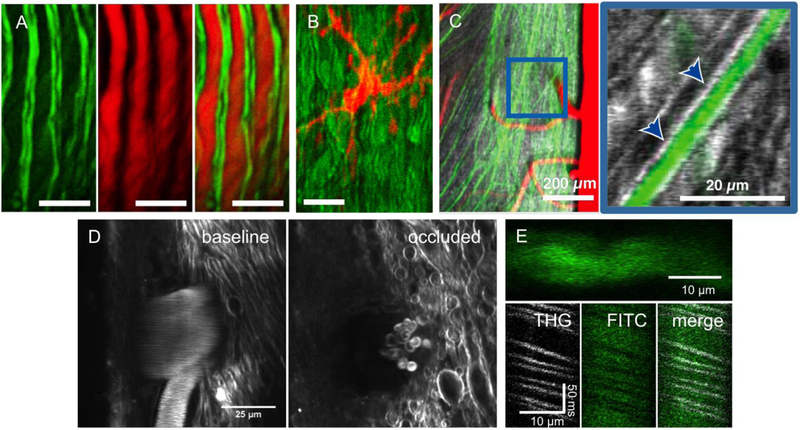
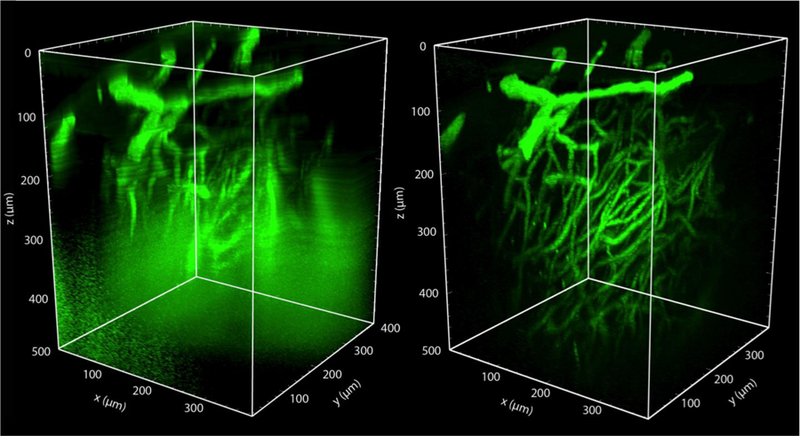
In vivo optical imaging has enabled detailed studies of cellular dynamics in the brain of rodents in both healthy and diseased states. Such studies were made possible by three advances: surgical preparations that give optical access to the brain; strategies for in vivo labeling of cells with structural and functional fluorescent indicators; and optical imaging techniques that are relatively insensitive to light scattering by tissue. In vivo imaging in the rodent spinal cord has lagged behind than that in the brain, largely due to the anatomy around the spinal cord that complicates the surgical preparation, and to the strong optical scattering of the dorsal white matter that limits the ability to image deep into the spinal cord. Here, we review recent advances in surgical methods, labeling strategies, and optical tools that have enabled in vivo, high-resolution imaging of the dynamic behaviors of cells in the spinal cord in mice. Surgical preparations that enable long-term optical access and robust stabilization of the spinal cord are now available. Labeling strategies that have been used in the spinal cord tend to follow those that have been used in the brain, and some recent advances in genetically-encoded labeling strategies remain to be capitalized on. The optical imaging methods used to date, including two photon excited fluorescence microscopy, are largely limited to imaging the superficial layers of the spinal cord by the optical scattering of the white matter. Finally, we show preliminary data that points to the use of higher-order nonlinear optical processes, such as three photon excited fluorescence, as a means to image deeper into the mouse spinal cord.
Keywords: Animal models; Chronic imaging; Intravital microscopy; Spinal cord; Two-photon excited fluorescence.
Copyright © 2019 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
