

HOME: a histogram based machine learning approach for effective identification of differentially methylated regions
- PMID: 31096906
- PMCID: PMC6521357
- DOI: 10.1186/s12859-019-2845-y
HOME: a histogram based machine learning approach for effective identification of differentially methylated regions
Abstract
Background: The development of whole genome bisulfite sequencing has made it possible to identify methylation differences at single base resolution throughout an entire genome. However, a persistent challenge in DNA methylome analysis is the accurate identification of differentially methylated regions (DMRs) between samples. Sensitive and specific identification of DMRs among different conditions requires accurate and efficient algorithms, and while various tools have been developed to tackle this problem, they frequently suffer from inaccurate DMR boundary identification and high false positive rate.
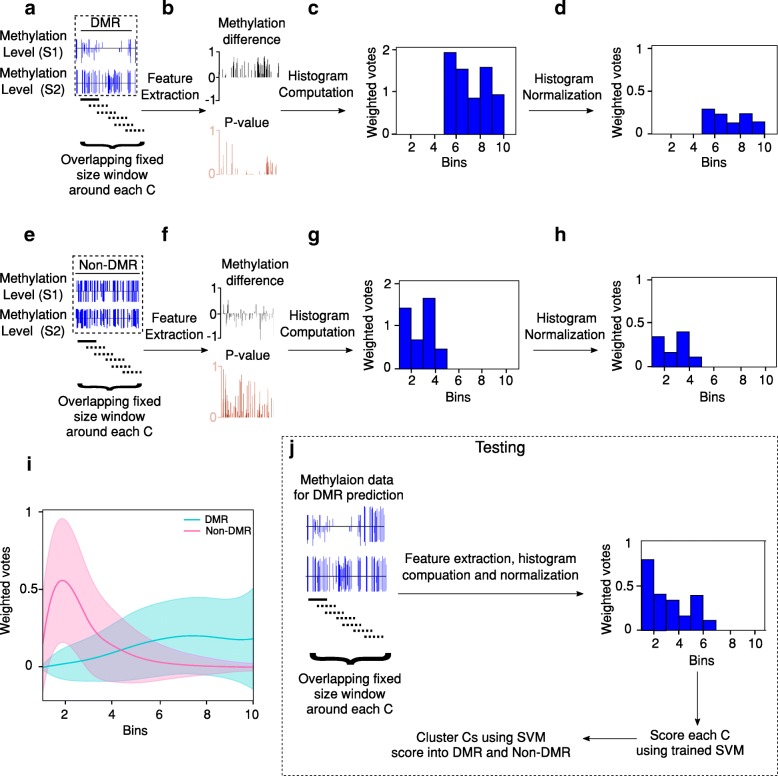
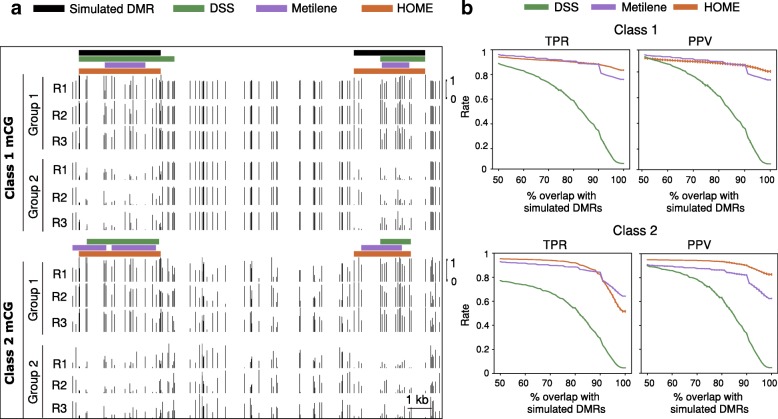
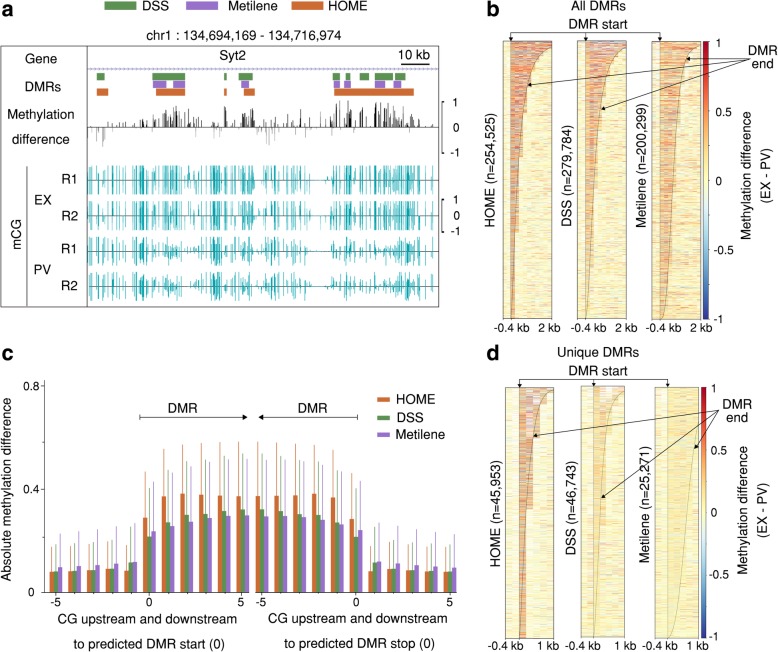
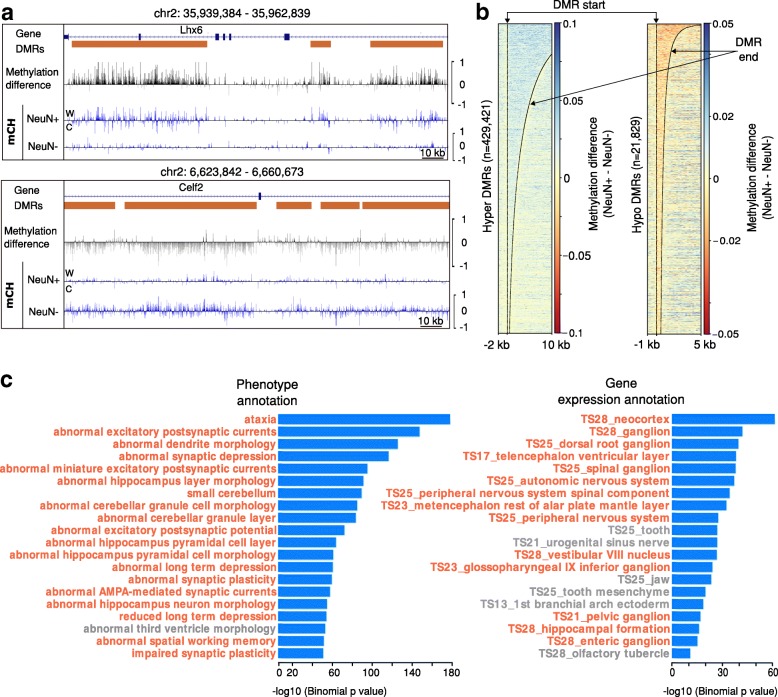
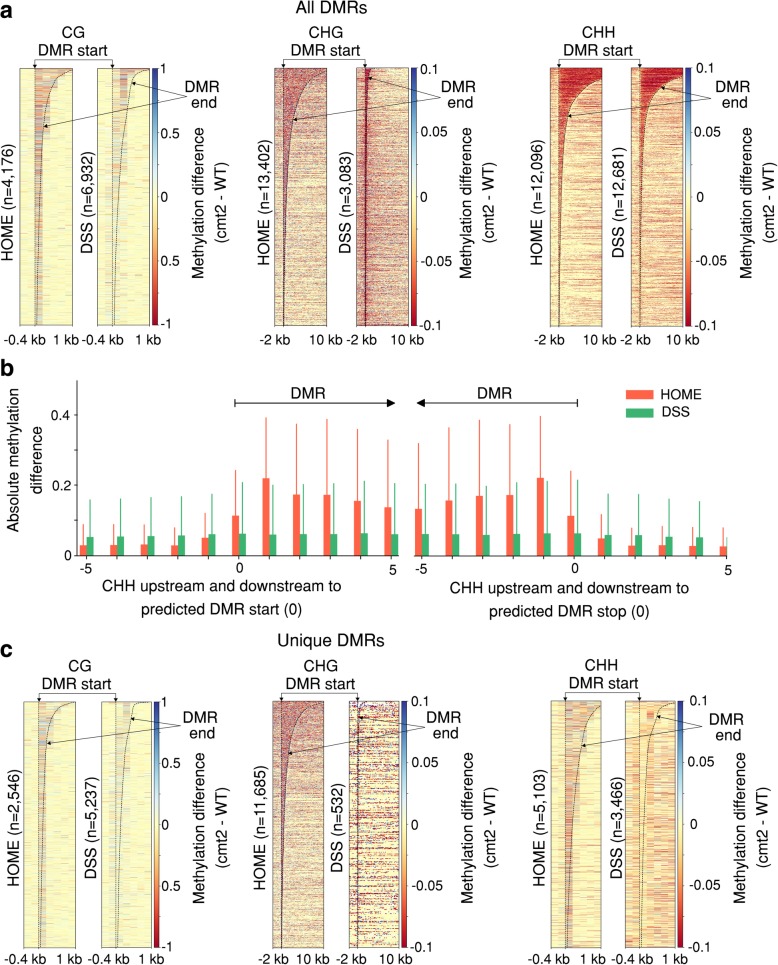
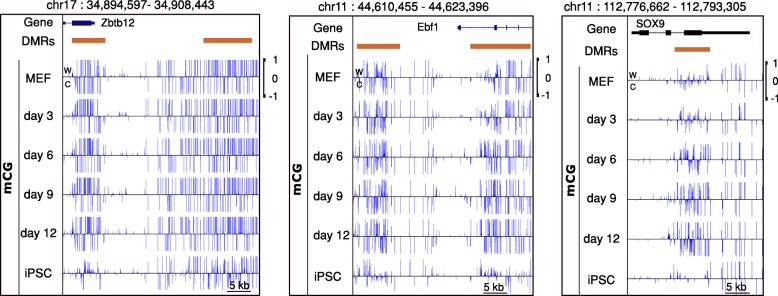
Results: We present a novel Histogram Of MEthylation (HOME) based method that takes into account the inherent difference in the distribution of methylation levels between DMRs and non-DMRs to discriminate between the two using a Support Vector Machine. We show that generated features used by HOME are dataset-independent such that a classifier trained on, for example, a mouse methylome training set of regions of differentially accessible chromatin, can be applied to any other organism's dataset and identify accurate DMRs. We demonstrate that DMRs identified by HOME exhibit higher association with biologically relevant genes, processes, and regulatory events compared to the existing methods. Moreover, HOME provides additional functionalities lacking in most of the current DMR finders such as DMR identification in non-CG context and time series analysis. HOME is freely available at https://github.com/ListerLab/HOME .
Conclusion: HOME produces more accurate DMRs than the current state-of-the-art methods on both simulated and biological datasets. The broad applicability of HOME to identify accurate DMRs in genomic data from any organism will have a significant impact upon expanding our knowledge of how DNA methylation dynamics affect cell development and differentiation.
Keywords: DMR identification; DNA methylation; Epigenetics; SVM; Whole genome bisulfite sequencing.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Richardson BC. Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J Nutr. 2002;132(8):2401S–2405S. - PubMed
-
- Khavari DA, Sen GL, Rinn JL. DNA methylation and epigenetic control of cellular differentiation. Cell Cycle. 2010;9(19):3880–3883. - PubMed
-
- Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
