

Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy
- PMID: 31097342
- PMCID: PMC6527362
- DOI: 10.1016/j.immuni.2019.04.010
Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy
Abstract
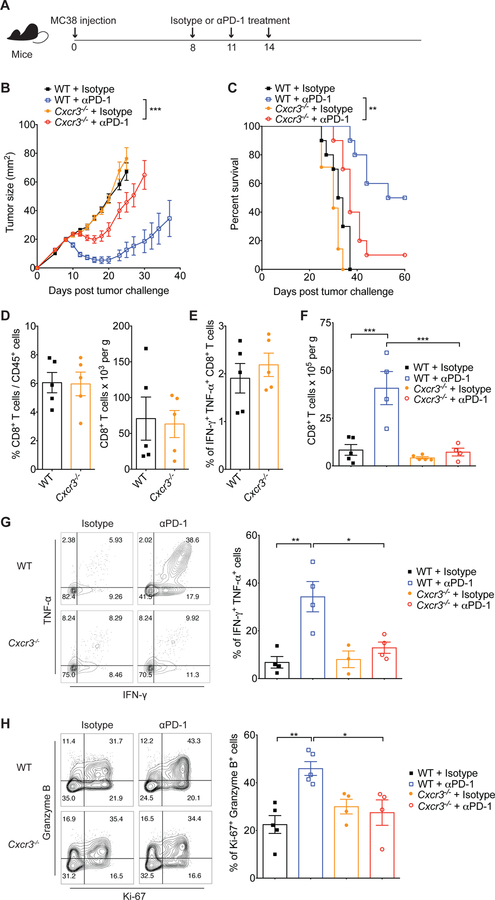
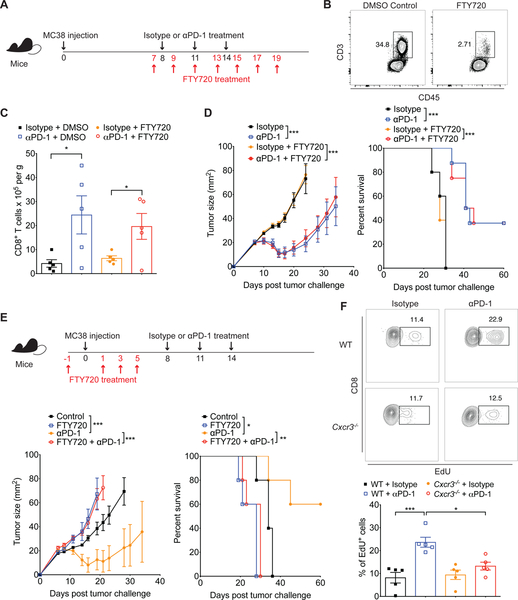
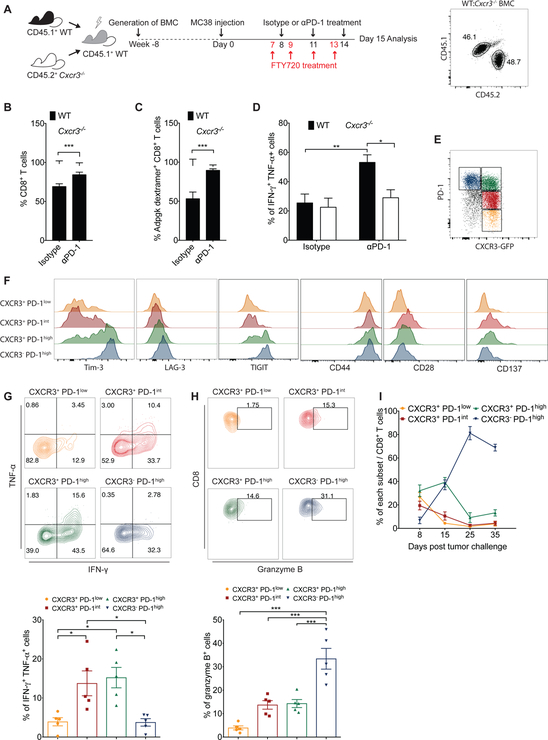
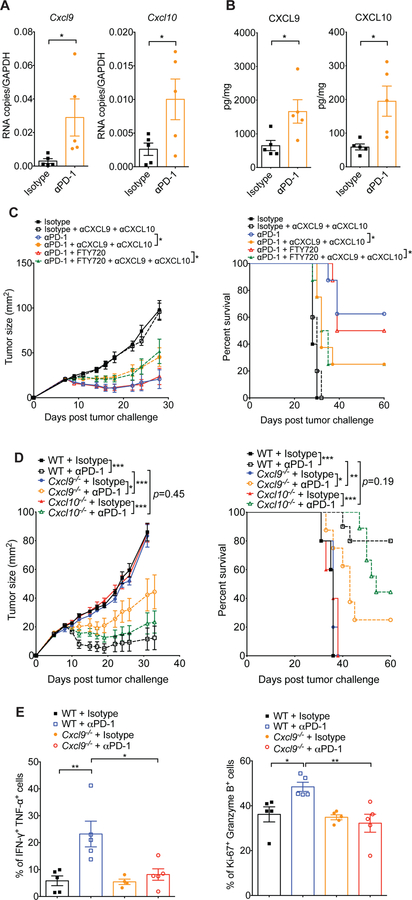
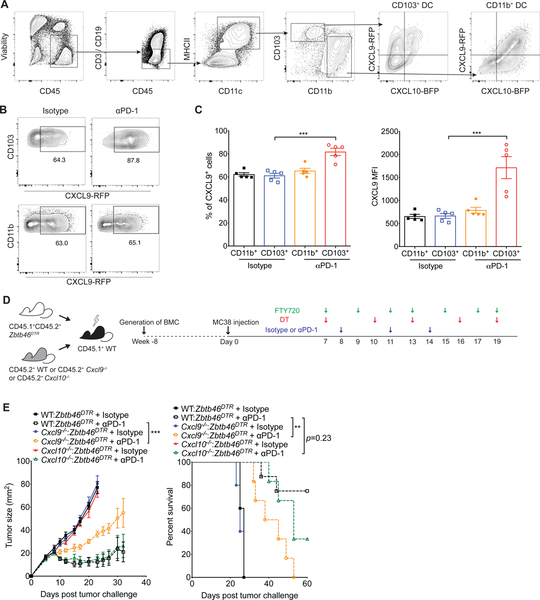
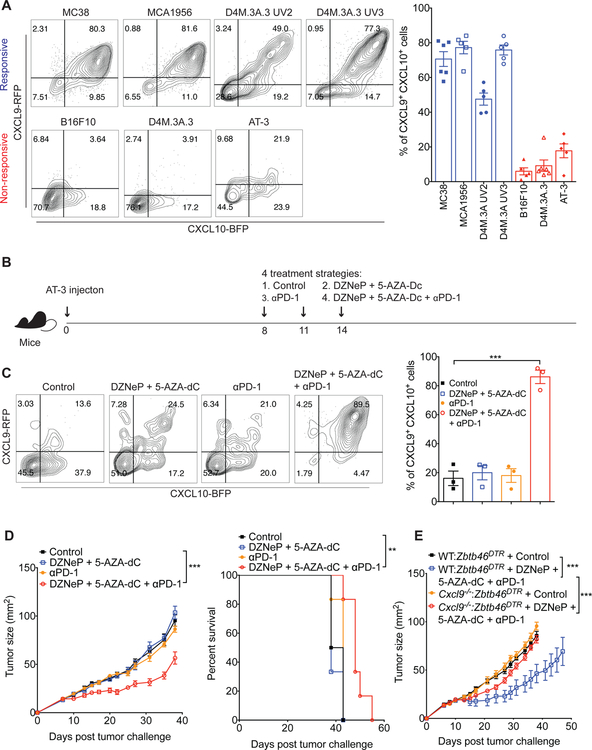
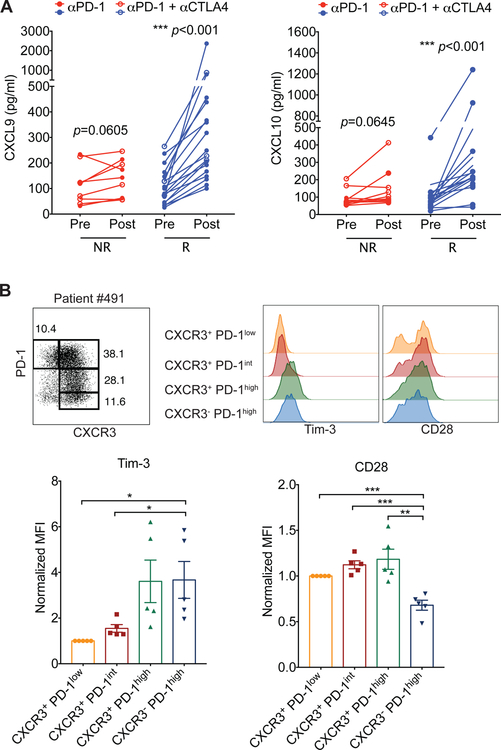
Despite compelling rates of durable clinical responses to programmed cell death-1 (PD-1) blockade, advances are needed to extend these benefits to resistant tumors. We found that tumor-bearing mice deficient in the chemokine receptor CXCR3 responded poorly to anti-PD-1 treatment. CXCR3 and its ligand CXCL9 were critical for a productive CD8+ T cell response in tumor-bearing mice treated with anti-PD-1 but were not required for the infiltration of CD8+ T cells into tumors. The anti-PD-1-induced anti-tumor response was facilitated by CXCL9 production from intratumoral CD103+ dendritic cells, suggesting that CXCR3 facilitates dendritic cell-T cell interactions within the tumor microenvironment. CXCR3 ligands in murine tumors and in plasma of melanoma patients were an indicator of clinical response to anti-PD-1, and their induction in non-responsive murine tumors promoted responsiveness to anti-PD-1. Our data suggest that the CXCR3 chemokine system is a biomarker for sensitivity to PD-1 blockade and that augmenting the intratumoral function of this chemokine system could improve clinical outcomes.
Keywords: CD8(+) T cells; CXCL10; CXCL9; CXCR3; PD-1; chemokine; dendritic cells; immune checkpoint; immunotherapy.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
A.D.L. has consulted for Merck, Lilly, Sanofi, Idera, Receptos, and Celtaxsys. D.E.F. has a financial interest in Soltego, Inc., a company developing SIK inhibitors for topical skin darkening treatments that might be used for a broad set of human applications. Dr. Fisher’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. G.J.F. has patents/pending royalties on the PD-1 pathway from Roche, Merck, Bristol-Myers-Squibb, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Dako and Novartis and has served on advisory boards for Roche, Bristol-Myers- Squibb, Xios, and Origimed.
Figures
Comment in
-
Predicting the anti-PD1 response.Nat Rev Immunol. 2019 Jul;19(7):414-415. doi: 10.1038/s41577-019-0184-x. Nat Rev Immunol. 2019. PMID: 31150011 No abstract available.
-
CXCR3-CXCL9: It's All in the Tumor.Immunity. 2019 Jun 18;50(6):1347-1349. doi: 10.1016/j.immuni.2019.05.013. Immunity. 2019. PMID: 31216458
References
-
- Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, Jacobs H, and Borst J (2017). CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity 47, 848–861. - PubMed
-
- Berghuis D, Santos SJ, Baelde HJ, Taminiau AH, Maarten Egeler R, Schilham MW, Hogendoorn PC, and Lankester AC (2011). Pro‐inflammatory chemokine-chemokine receptor interactions within the Ewing sarcoma microenvironment determine CD8+ T‐lymphocyte infiltration and affect tumour progression. J. Pathol 223, 347–357. - PubMed
-
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. - PubMed
-
- Christen U, McGavern DB, Luster AD, von Herrath MG, and Oldstone MB (2003). Among CXCR3 chemokines, IFN-γ-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) 10) but not monokine induced by IFN-γ (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J. Immunol 171, 6838–6845. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
