

TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: transcriptomic and epigenomic analysis
- PMID: 31097419
- PMCID: PMC6692909
- DOI: 10.1136/annrheumdis-2018-214783
TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: transcriptomic and epigenomic analysis
Abstract
Objective: We investigated genome-wide changes in gene expression and chromatin remodelling induced by tumour necrosis factor (TNF) in fibroblast-like synoviocytes (FLS) and macrophages to better understand the contribution of FLS to the pathogenesis of rheumatoid arthritis (RA).
Methods: FLS were purified from patients with RA and CD14+ human monocyte-derived macrophages were obtained from healthy donors. RNA-sequencing, histone 3 lysine 27 acetylation (H3K27ac), chromatin immunoprecipitation-sequencing (ChIP-seq) and assay for transposable accessible chromatin by high throughput sequencing (ATAC-seq) were performed in control and TNF-stimulated cells.
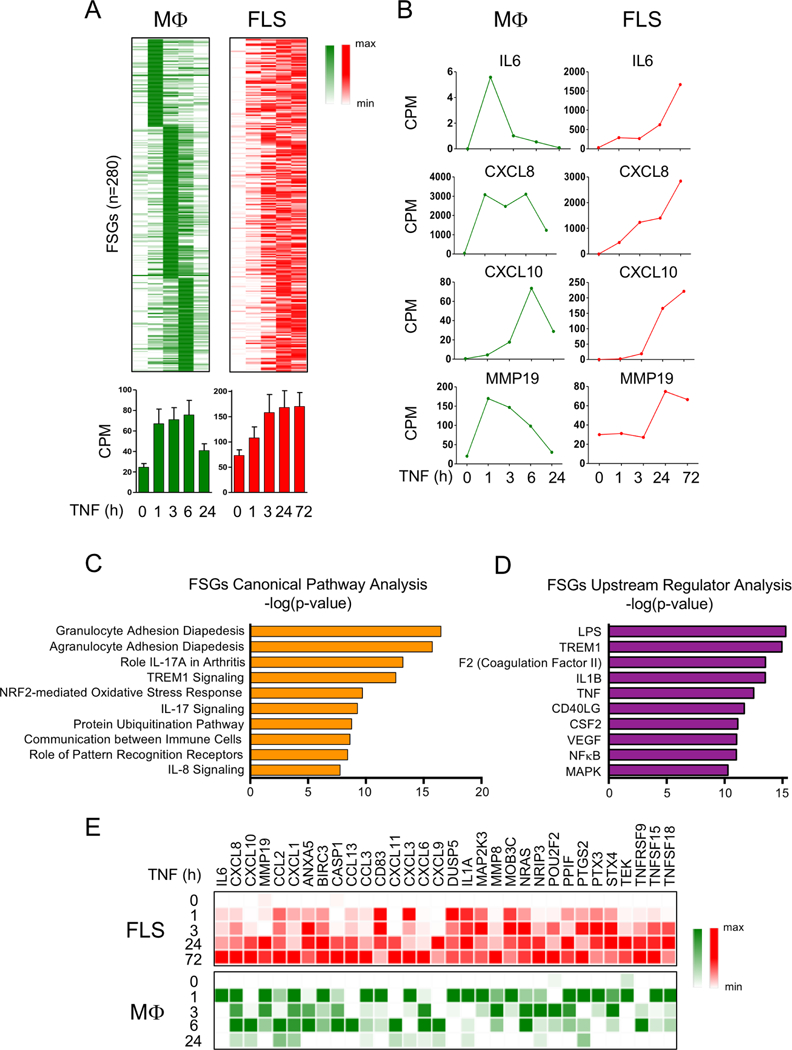
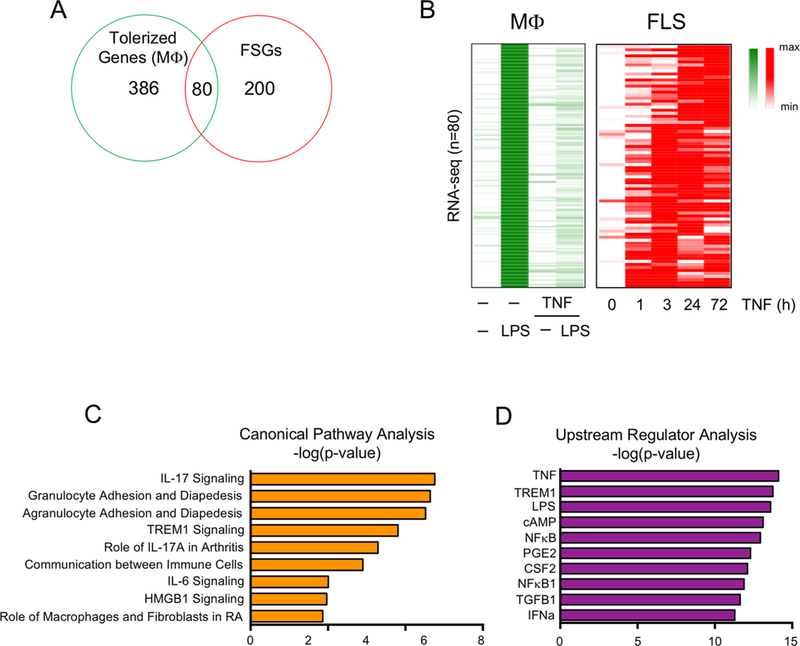
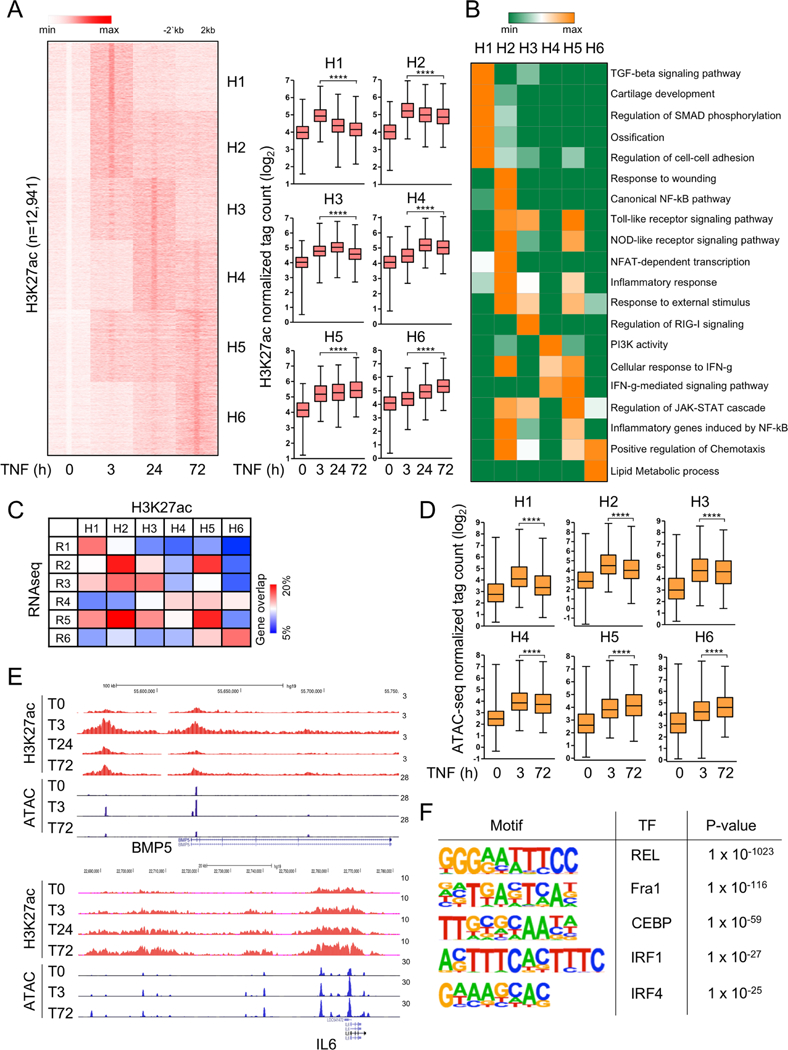
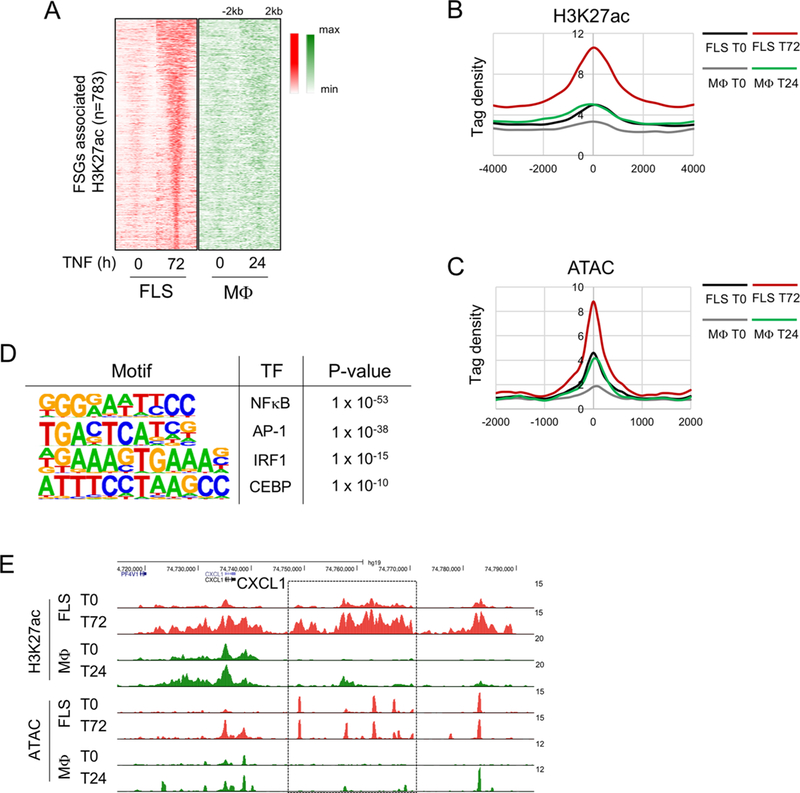
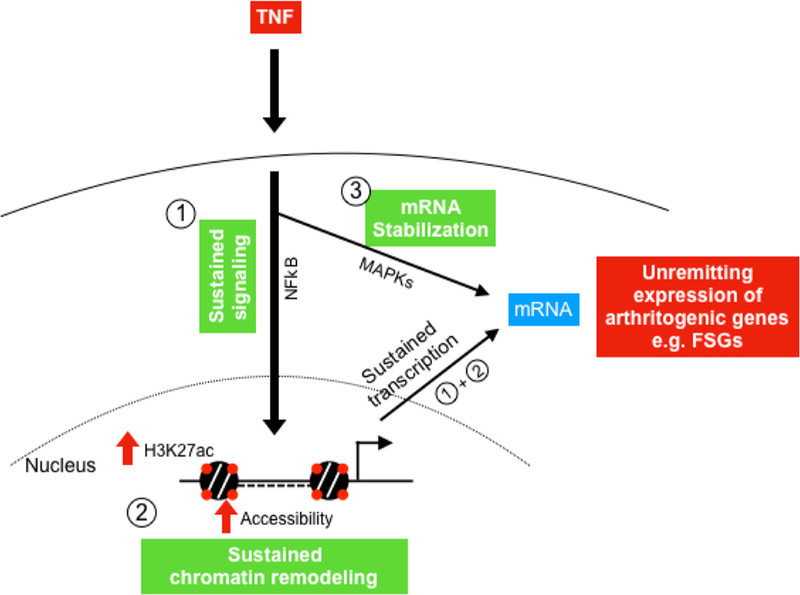
Results: We discovered 280 TNF-inducible arthritogenic genes which are transiently expressed and subsequently repressed in macrophages, but in RA, FLS are expressed with prolonged kinetics that parallel the unremitting kinetics of RA synovitis. 80 out of these 280 fibroblast-sustained genes (FSGs) that escape repression in FLS relative to macrophages were desensitised (tolerised) in macrophages. Epigenomic analysis revealed persistent H3K27 acetylation and increased chromatin accessibility in regulatory elements associated with FSGs in TNF-stimulated FLS. The accessible regulatory elements of FSGs were enriched in binding motifs for nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), interferon-regulatory factors (IRFs) and activating protein-1 (AP-1). Inhibition of bromodomain and extra-terminal motif (BET) proteins, which interact with histone acetylation, suppressed sustained induction of FSGs by TNF.
Conclusion: Our genome-wide analysis has identified the escape of genes from transcriptional repression in FLS as a novel mechanism potentially contributing to the chronic unremitting synovitis observed in RA. Our finding that TNF induces sustained chromatin activation in regulatory elements of the genes that escape repression in RA FLS suggests that altering or targeting chromatin states in FLS (eg, with inhibitors of BET proteins) is an attractive therapeutic strategy.
Keywords: fibroblasts; rheumatoid arthritis; synovitis; tnf-alpha.
© Author(s) (or their employer(s)) 2019. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared. GDK is at the time of publication of this study an employee at Regeneron Pharmaceuticals Inc. and declares no conflict of interest related to the content of this manuscript.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
