

Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades
- PMID: 31097702
- PMCID: PMC6522487
- DOI: 10.1038/s41467-019-10191-3
Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades
Abstract
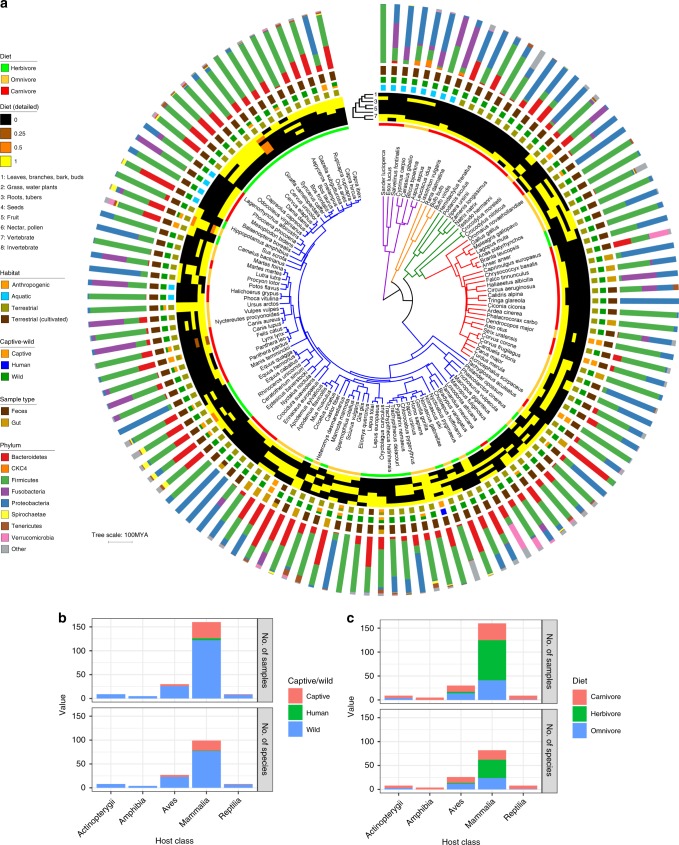
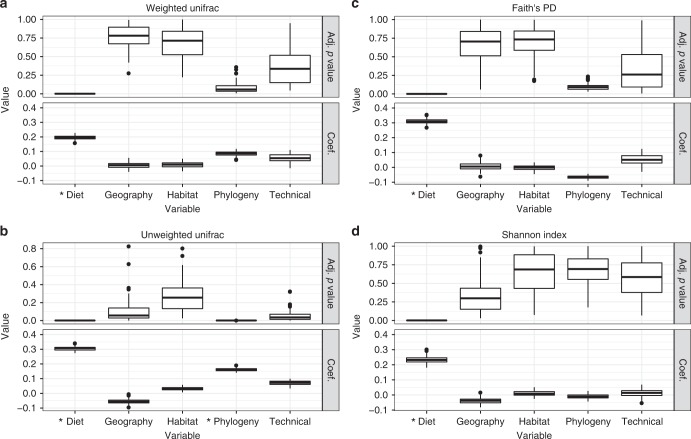
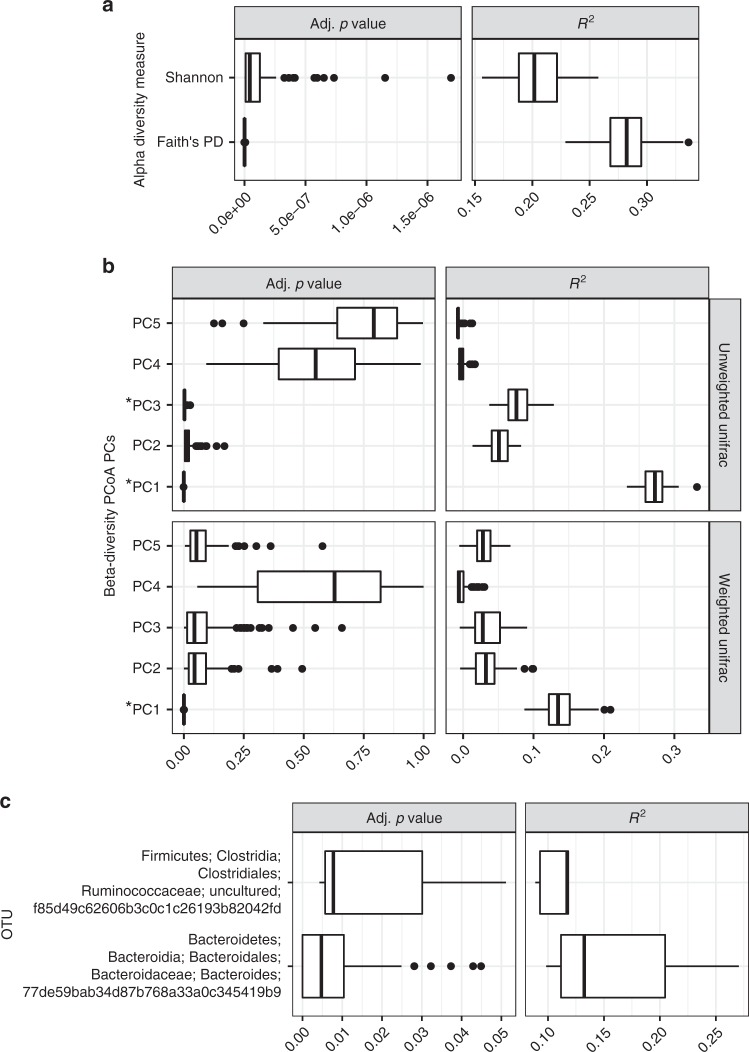
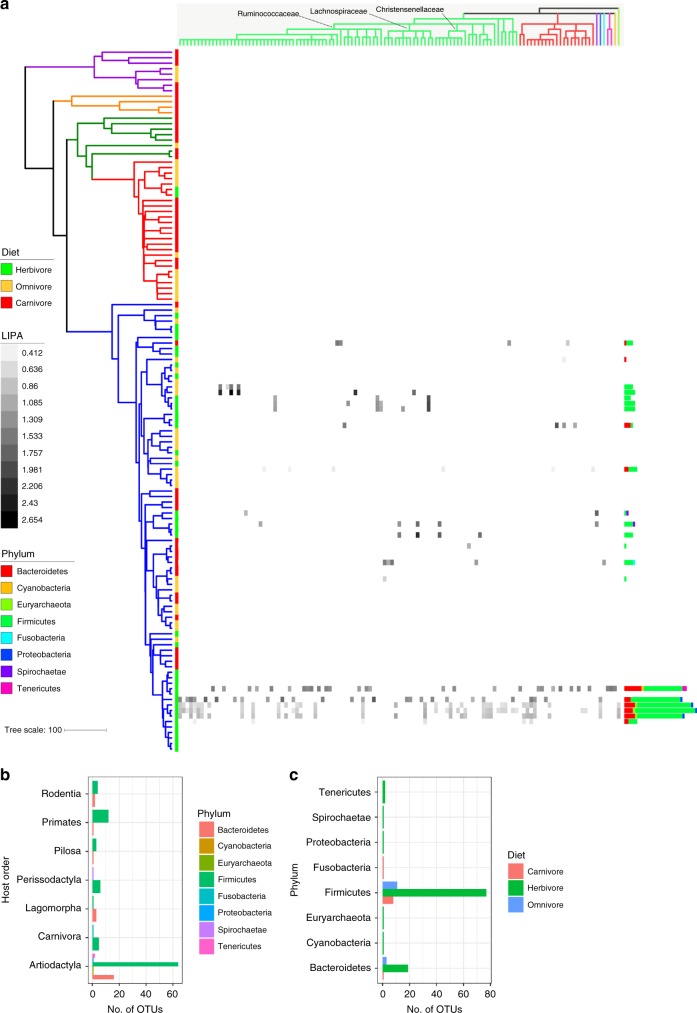
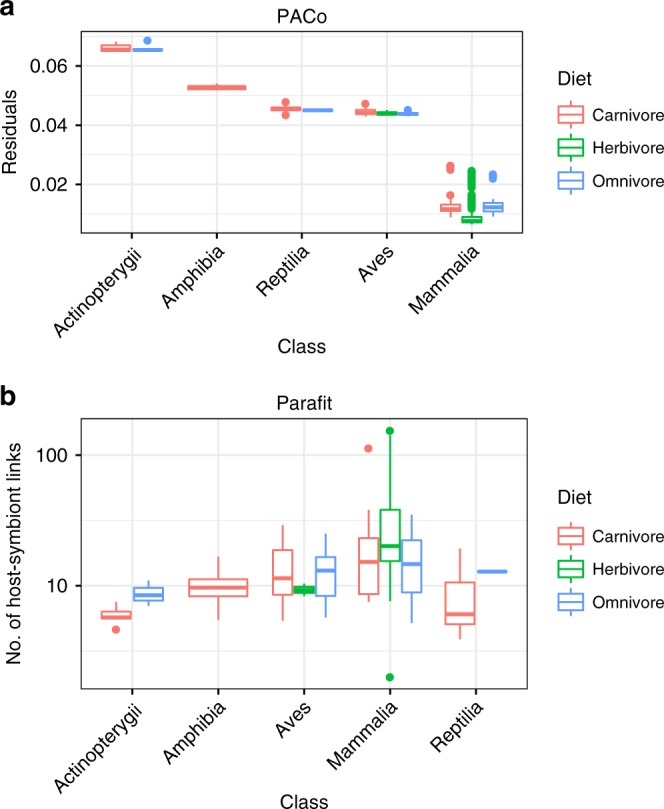
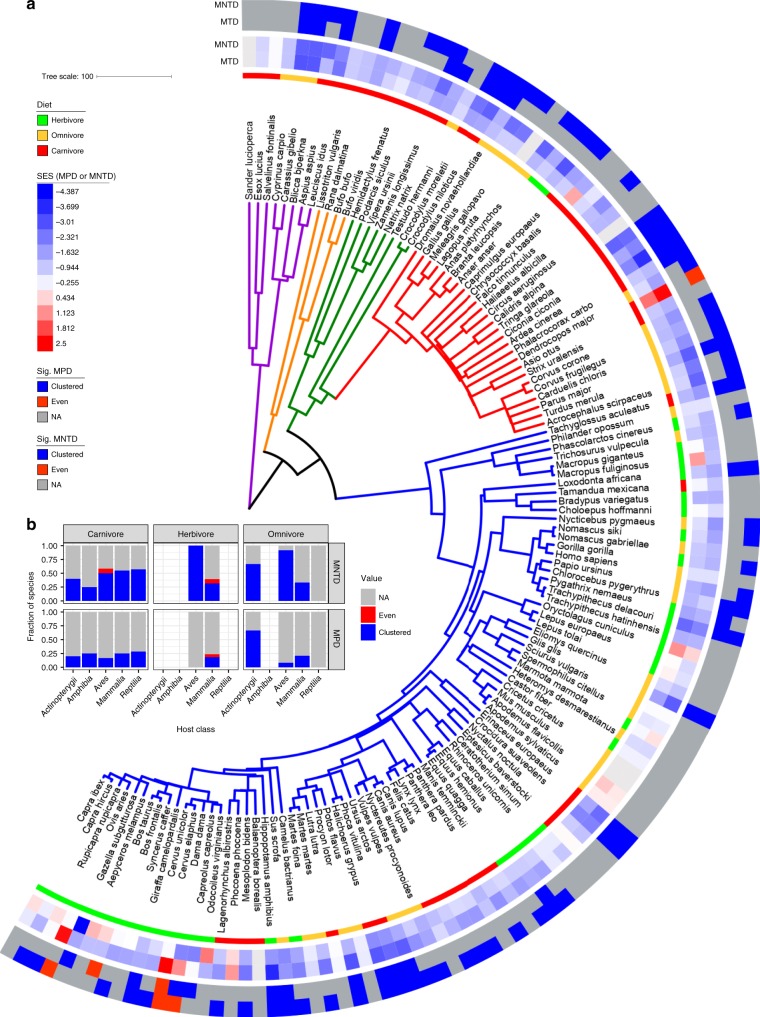
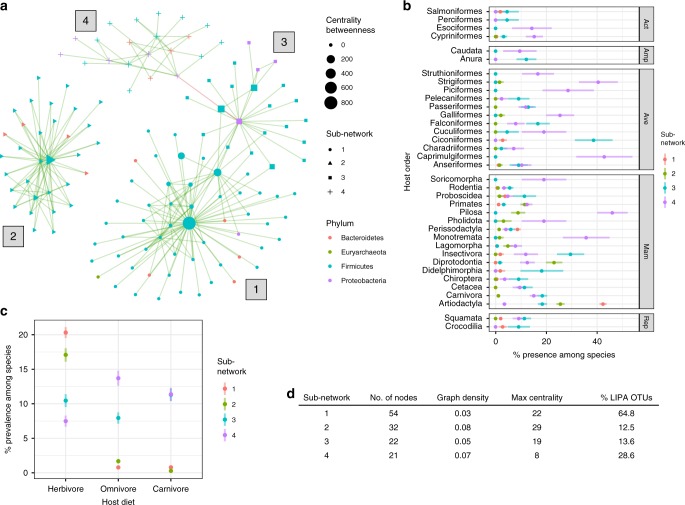
Multiple factors modulate microbial community assembly in the vertebrate gut, though studies disagree as to their relative contribution. One cause may be a reliance on captive animals, which can have very different gut microbiomes compared to their wild counterparts. To resolve this disagreement, we analyze a new, large, and highly diverse animal distal gut 16 S rRNA microbiome dataset, which comprises 80% wild animals and includes members of Mammalia, Aves, Reptilia, Amphibia, and Actinopterygii. We decouple the effects of host evolutionary history and diet on gut microbiome diversity and show that each factor modulates different aspects of diversity. Moreover, we resolve particular microbial taxa associated with host phylogeny or diet and show that Mammalia have a stronger signal of cophylogeny. Finally, we find that environmental filtering and microbe-microbe interactions differ among host clades. These findings provide a robust assessment of the processes driving microbial community assembly in the vertebrate intestine.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
