

Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo
- PMID: 31097717
- PMCID: PMC6522502
- DOI: 10.1038/s41467-019-09963-8
Innate lymphocyte-induced CXCR3B-mediated melanocyte apoptosis is a potential initiator of T-cell autoreactivity in vitiligo
Abstract
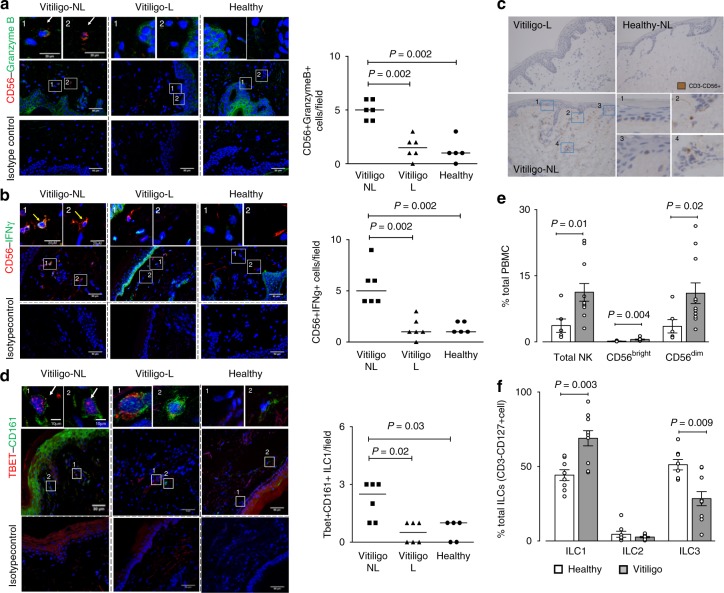
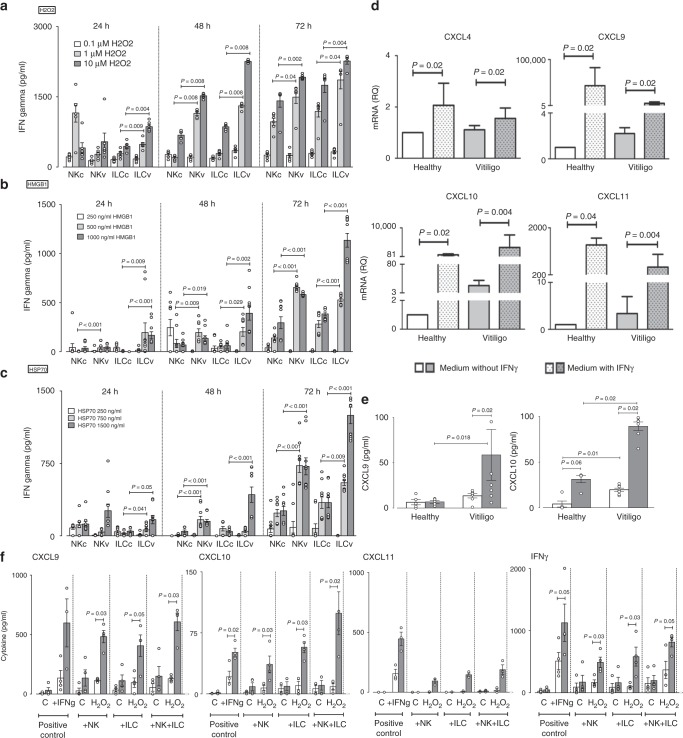
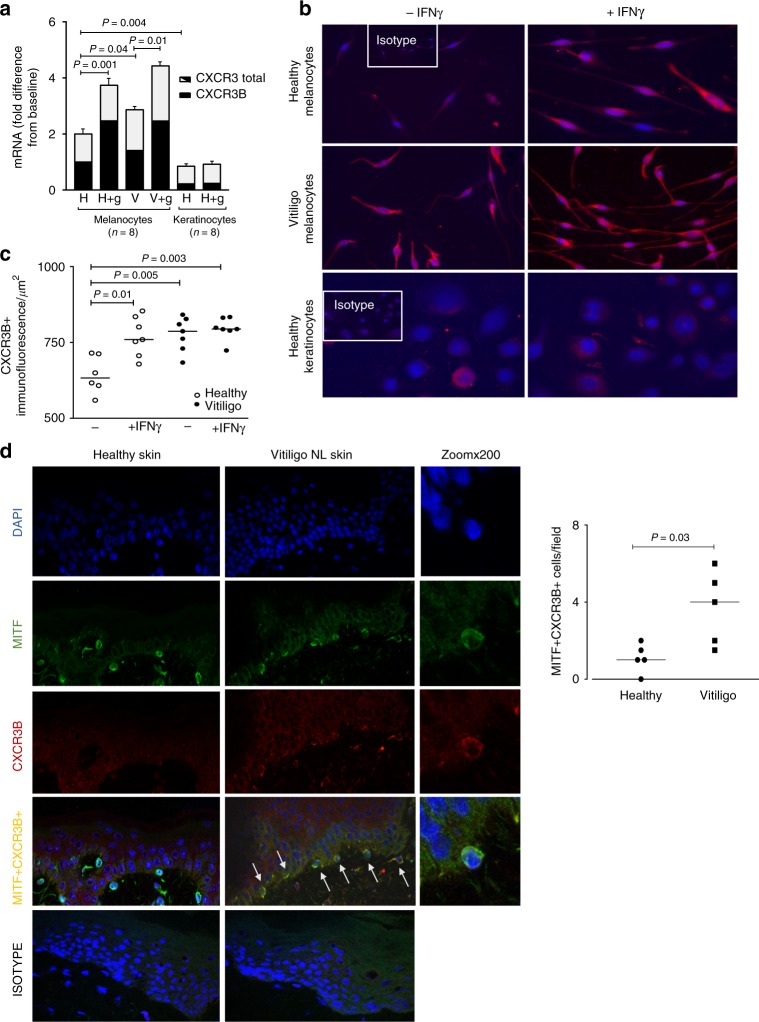
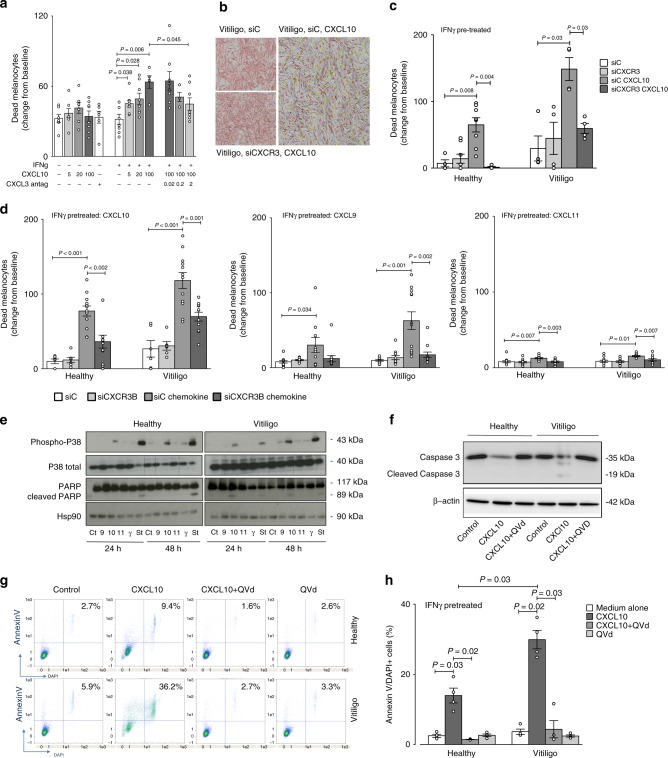
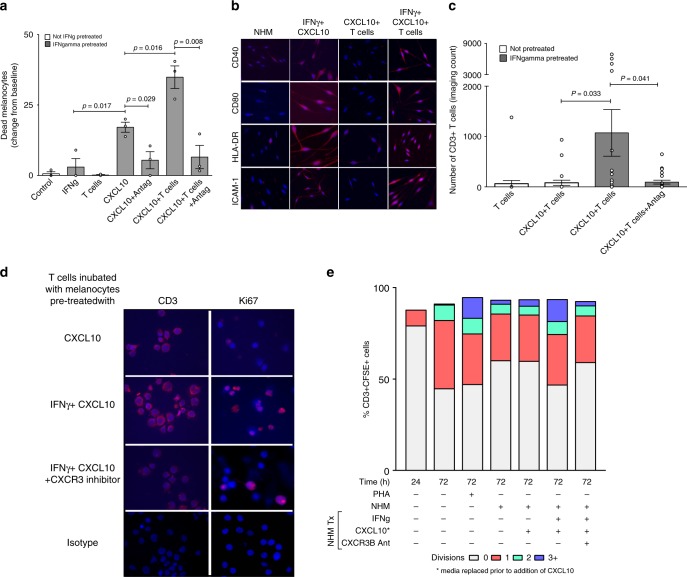
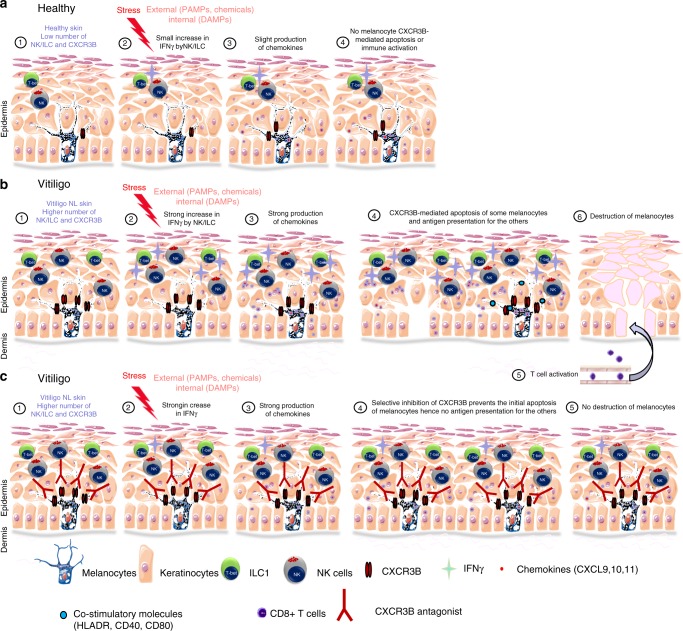
T-cells play a crucial role in progression of autoimmunity, including vitiligo, yet the initial steps triggering their activation and tissue damage remain unknown. Here we demonstrate increased presence of type-1 innate lymphoid cells (NK and ILC1)-producing interferon gamma (IFNγ) in the blood and in non-lesional skin of vitiligo patients. Melanocytes of vitiligo patients have strong basal expression of chemokine-receptor-3 (CXCR3) isoform B which is directly regulated by IFNγ. CXCR3B activation by CXCL10 at the surface of cultured human melanocytes induces their apoptosis. The remaining melanocytes, activated by the IFNγ production, express co-stimulatory markers which trigger T-cell proliferation and subsequent anti-melanocytic immunity. Inhibiting the CXCR3B activation prevents this apoptosis and the further activation of T cells. Our results emphasize the key role of CXCR3B in apoptosis of melanocytes and identify CXCR3B as a potential target to prevent and to treat vitiligo by acting at the early stages of melanocyte destruction.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
