

Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight
- PMID: 31104630
- PMCID: PMC6526621
- DOI: 10.1186/s13059-019-1707-2
Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight
Abstract
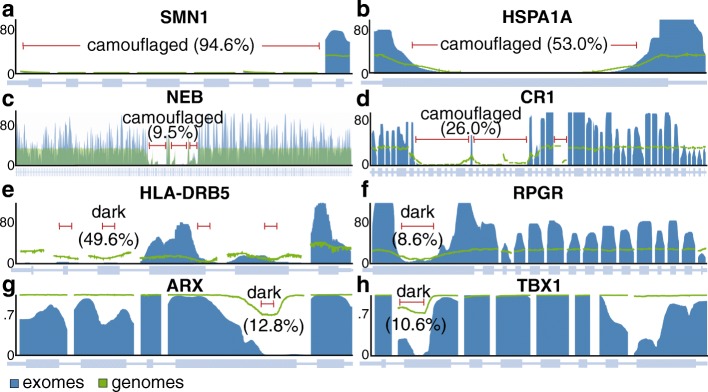
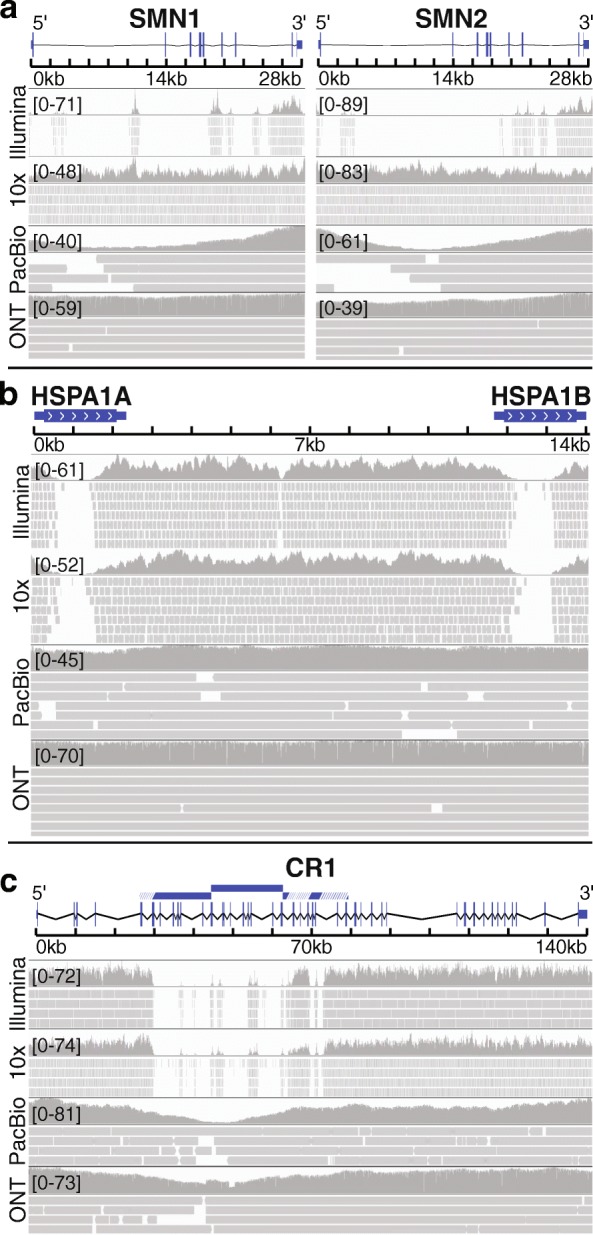
Background: The human genome contains "dark" gene regions that cannot be adequately assembled or aligned using standard short-read sequencing technologies, preventing researchers from identifying mutations within these gene regions that may be relevant to human disease. Here, we identify regions with few mappable reads that we call dark by depth, and others that have ambiguous alignment, called camouflaged. We assess how well long-read or linked-read technologies resolve these regions.
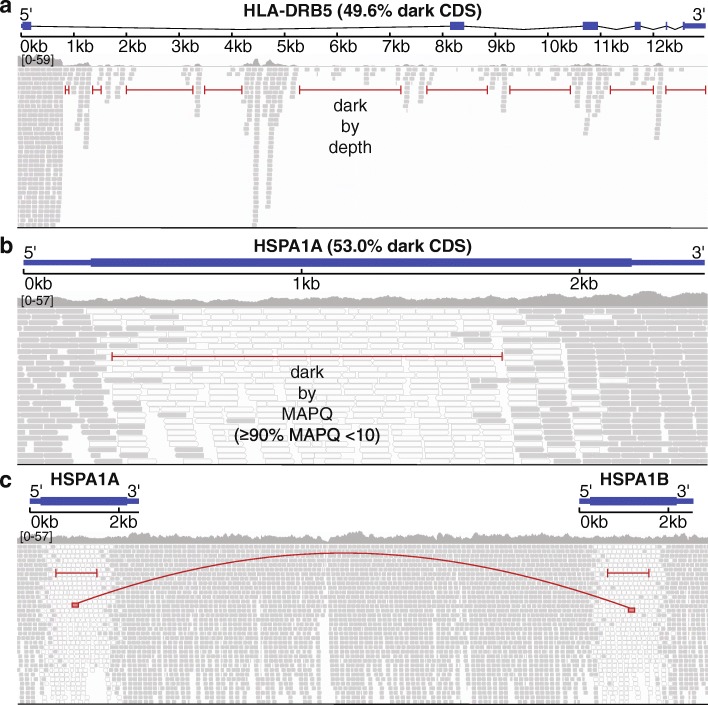
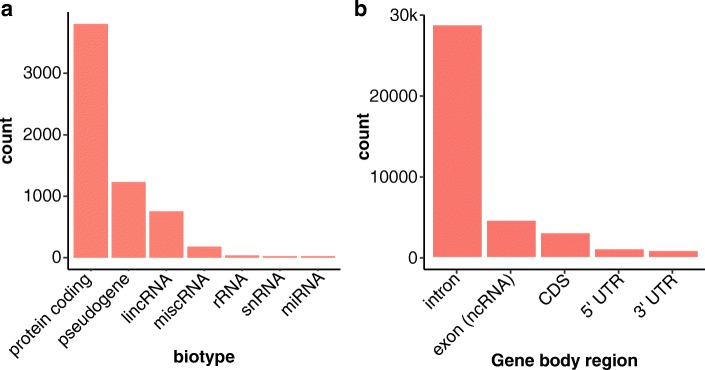
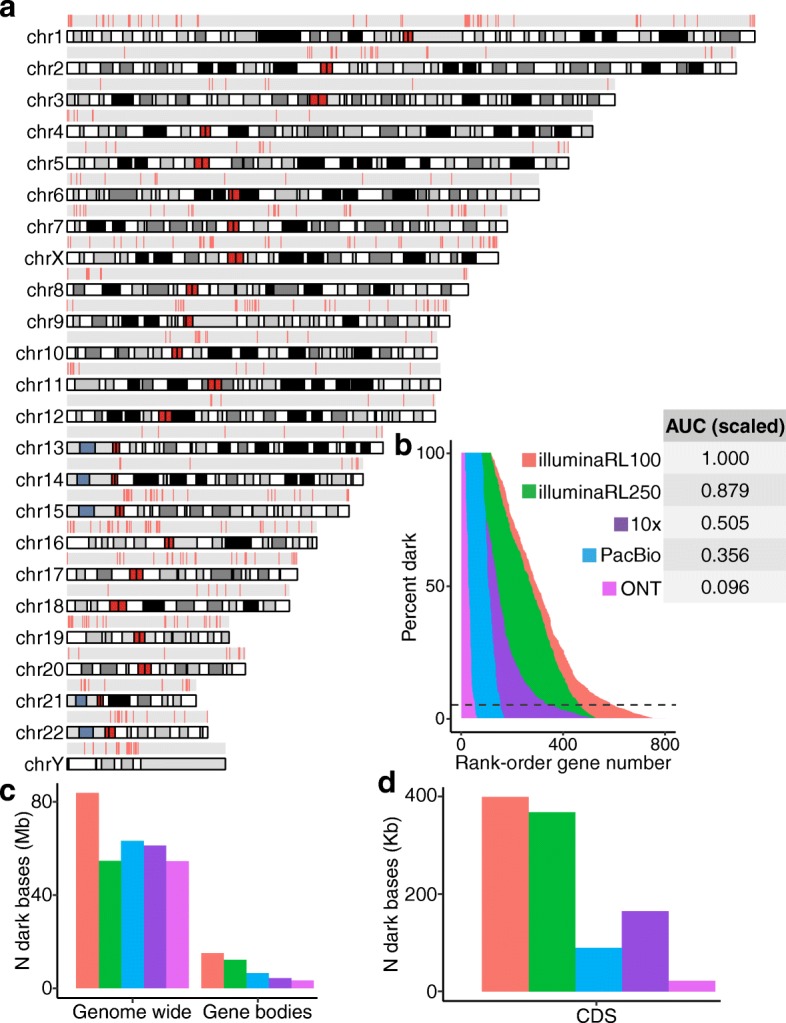
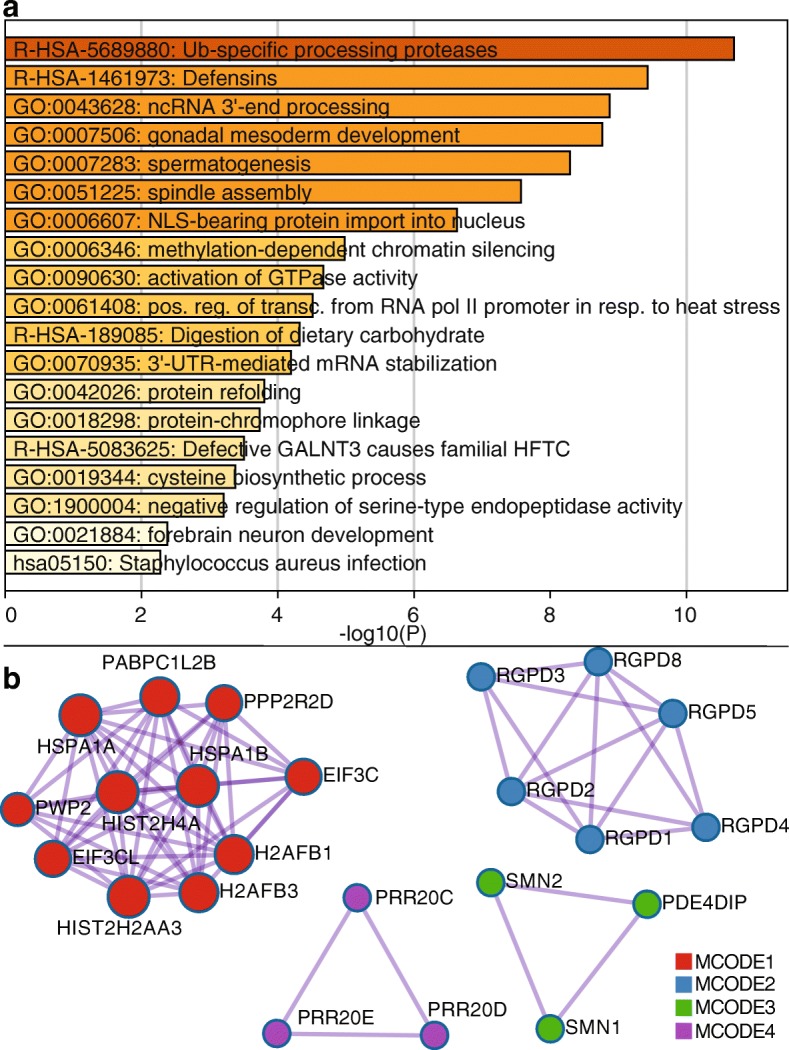
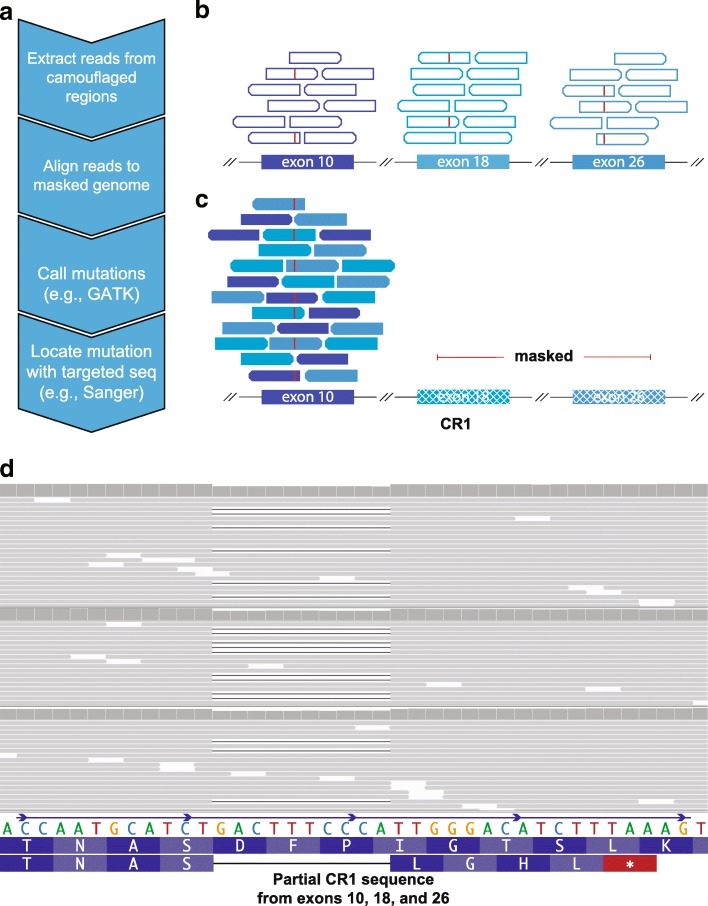
Results: Based on standard whole-genome Illumina sequencing data, we identify 36,794 dark regions in 6054 gene bodies from pathways important to human health, development, and reproduction. Of these gene bodies, 8.7% are completely dark and 35.2% are ≥ 5% dark. We identify dark regions that are present in protein-coding exons across 748 genes. Linked-read or long-read sequencing technologies from 10x Genomics, PacBio, and Oxford Nanopore Technologies reduce dark protein-coding regions to approximately 50.5%, 35.6%, and 9.6%, respectively. We present an algorithm to resolve most camouflaged regions and apply it to the Alzheimer's Disease Sequencing Project. We rescue a rare ten-nucleotide frameshift deletion in CR1, a top Alzheimer's disease gene, found in disease cases but not in controls.
Conclusions: While we could not formally assess the association of the CR1 frameshift mutation with Alzheimer's disease due to insufficient sample-size, we believe it merits investigating in a larger cohort. There remain thousands of potentially important genomic regions overlooked by short-read sequencing that are largely resolved by long-read technologies.
Keywords: 10x Genomics; APOE; Alzheimer’s Disease Sequencing Project (ADSP); CR1; Camouflaged genes; Dark genes; Long-read sequencing; Oxford Nanopore Technologies (ONT); Pacific Biosciences (PacBio).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, et al. Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol Neurodegener. 2018;13:46. doi: 10.1186/s13024-018-0274-4. - DOI - PMC - PubMed
-
- Callaway E. Human brain shaped by duplicate genes. Nature. 2012. 10.1038/nature.2012.10584.
Publication types
MeSH terms
Grants and funding
- RF1 AG062077/AG/NIA NIH HHS/United States
- N01 HC085081/HL/NHLBI NIH HHS/United States
- U01 AG049505/AG/NIA NIH HHS/United States
- HHSN268201100012C/HL/NHLBI NIH HHS/United States
- RC2 HL102419/HL/NHLBI NIH HHS/United States
- R35 NS097273/NS/NINDS NIH HHS/United States
- HHSN268201100009I/HL/NHLBI NIH HHS/United States
- N01 HC085080/HL/NHLBI NIH HHS/United States
- U01 HL096812/HL/NHLBI NIH HHS/United States
- U54 AG052427/AG/NIA NIH HHS/United States
- R01 NS017950/NS/NINDS NIH HHS/United States
- R21 AG047327/AG/NIA NIH HHS/United States
- R01 AG054076/AG/NIA NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- NS097261/NS/NINDS NIH HHS/United States
- HHSN268201100010C/HL/NHLBI NIH HHS/United States
- AG047327/AG/NIA NIH HHS/United States
- NS084974/NS/NINDS NIH HHS/United States
- AG049992/AG/NIA NIH HHS/United States
- R01 AG015928/AG/NIA NIH HHS/United States
- U24 AG021886/AG/NIA NIH HHS/United States
- HHSN268201100008C/HL/NHLBI NIH HHS/United States
- U01 HL080295/HL/NHLBI NIH HHS/United States
- HHSN268201500001C/HL/NHLBI NIH HHS/United States
- NS094137/NS/NINDS NIH HHS/United States
- U24 AG072122/AG/NIA NIH HHS/United States
- U01 AG049507/AG/NIA NIH HHS/United States
- U01 HL096917/HL/NHLBI NIH HHS/United States
- HHSN268201100008I/HL/NHLBI NIH HHS/United States
- HHSN268201100005G/HL/NHLBI NIH HHS/United States
- N01 HC085082/HL/NHLBI NIH HHS/United States
- U01 AG052411/AG/NIA NIH HHS/United States
- U01 AG032984/AG/NIA NIH HHS/United States
- U01 HL130114/HL/NHLBI NIH HHS/United States
- HHSN268201100007C/HL/NHLBI NIH HHS/United States
- HHSN268200800007C/HL/NHLBI NIH HHS/United States
- R35 NS097261/NS/NINDS NIH HHS/United States
- N01 HC085086/HL/NHLBI NIH HHS/United States
- N01 HC085083/HL/NHLBI NIH HHS/United States
- I 904/FWF_/Austrian Science Fund FWF/Austria
- NS093865/NS/NINDS NIH HHS/United States
- HHSN268201100011I/HL/NHLBI NIH HHS/United States
- HHSN268201100011C/HL/NHLBI NIH HHS/United States
- U01 AG016976/AG/NIA NIH HHS/United States
- U01 HL096902/HL/NHLBI NIH HHS/United States
- R01 NS093865/NS/NINDS NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- R01 AG049607/AG/NIA NIH HHS/United States
- UF1 AG047133/AG/NIA NIH HHS/United States
- U01 AG057659/AG/NIA NIH HHS/United States
- R01 HL105756/HL/NHLBI NIH HHS/United States
- RF1 AG051504/AG/NIA NIH HHS/United States
- R03 AG049992/AG/NIA NIH HHS/United States
- NS097273/NS/NINDS NIH HHS/United States
- HHSN268201100006C/HL/NHLBI NIH HHS/United States
- U24 AG041689/AG/NIA NIH HHS/United States
- HHSN268201200036C/HL/NHLBI NIH HHS/United States
- N01 HC025195/HL/NHLBI NIH HHS/United States
- R01 AG033193/AG/NIA NIH HHS/United States
- R01 AG061796/AG/NIA NIH HHS/United States
- N01 HC055222/HL/NHLBI NIH HHS/United States
- R01 NS088689/NS/NINDS NIH HHS/United States
- HHSN268201100005I/HL/NHLBI NIH HHS/United States
- HHSN268201500001I/HL/NHLBI NIH HHS/United States
- U01 AG049508/AG/NIA NIH HHS/United States
- NS084528/NS/NINDS NIH HHS/United States
- N01 HC085079/HL/NHLBI NIH HHS/United States
- U01 AG052410/AG/NIA NIH HHS/United States
- U01 HL096814/HL/NHLBI NIH HHS/United States
- R01 AG033040/AG/NIA NIH HHS/United States
- U54 HG003079/HG/NHGRI NIH HHS/United States
- P01 NS084974/NS/NINDS NIH HHS/United States
- U01 AG052409/AG/NIA NIH HHS/United States
- U01 AG046139/AG/NIA NIH HHS/United States
- R01 AG020098/AG/NIA NIH HHS/United States
- R21 NS084528/NS/NINDS NIH HHS/United States
- NS088689/NS/NINDS NIH HHS/United States
- NS099114/NS/NINDS NIH HHS/United States
- HHSN268201100009C/HL/NHLBI NIH HHS/United States
- R01 HL070825/HL/NHLBI NIH HHS/United States
- HHSN268201100005C/HL/NHLBI NIH HHS/United States
- U01 HL096899/HL/NHLBI NIH HHS/United States
- HHSN268201100007I/HL/NHLBI NIH HHS/United States
- P01 NS099114/NS/NINDS NIH HHS/United States
- U01 AG049506/AG/NIA NIH HHS/United States
- R01 NS094137/NS/NINDS NIH HHS/United States
- R01 AG023629/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous
