

Identification of a DNA N6-Adenine Methyltransferase Complex and Its Impact on Chromatin Organization
- PMID: 31104845
- PMCID: PMC6570567
- DOI: 10.1016/j.cell.2019.04.028
Identification of a DNA N6-Adenine Methyltransferase Complex and Its Impact on Chromatin Organization
Abstract
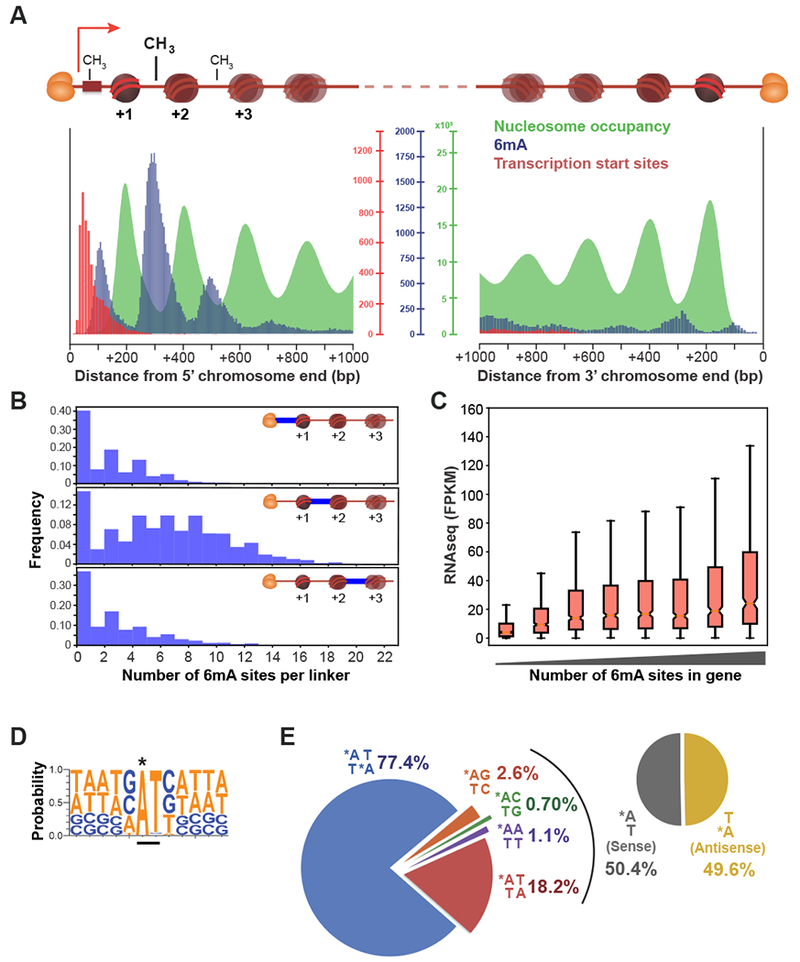
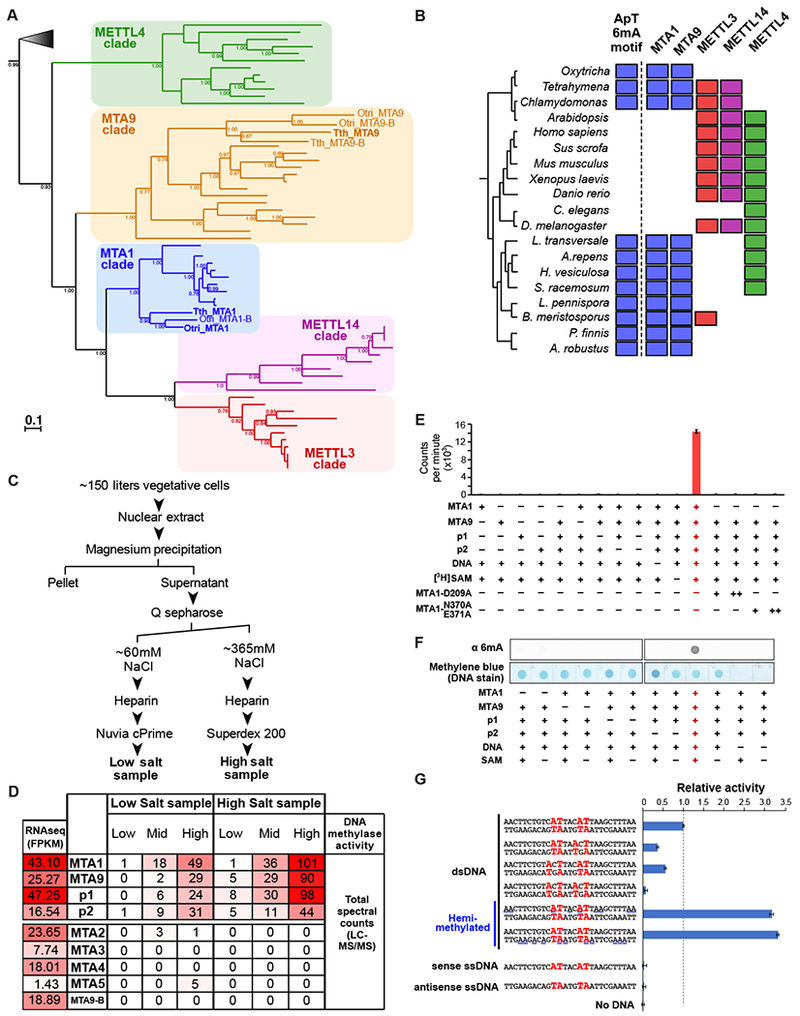
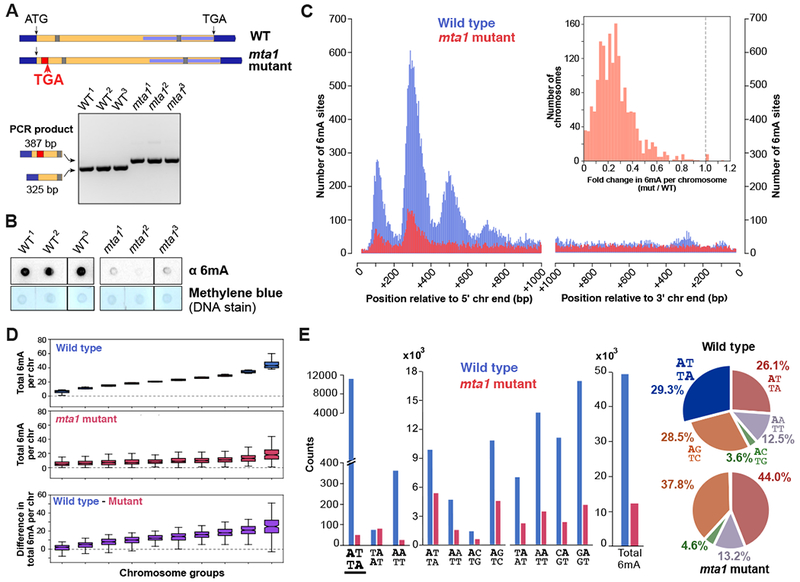
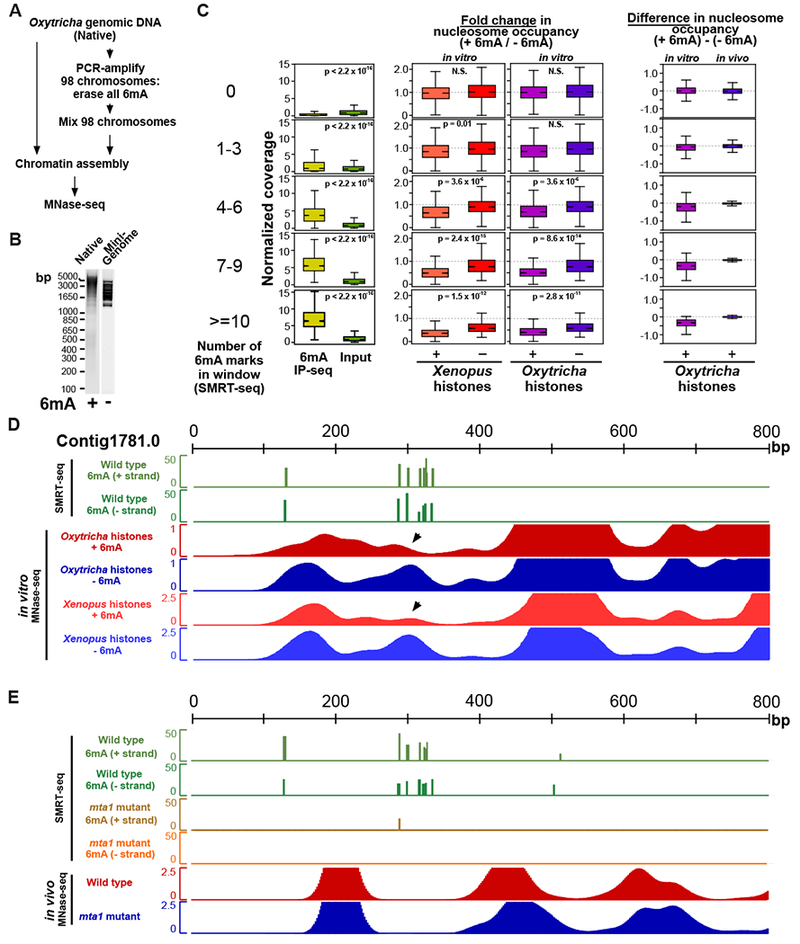
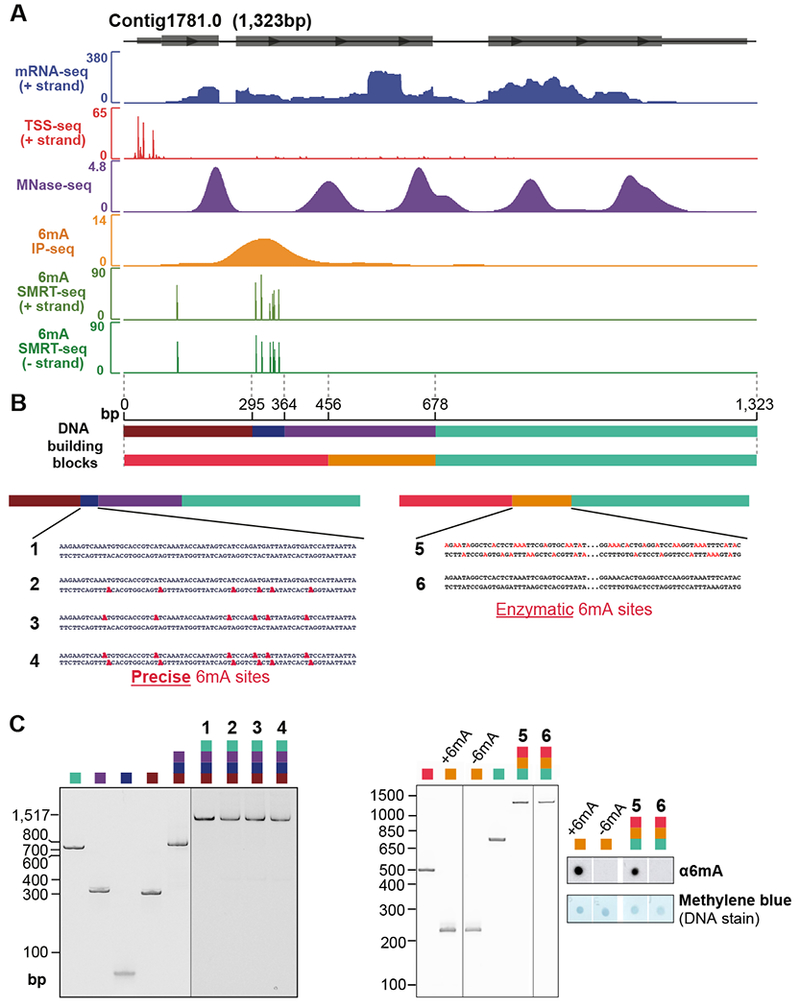
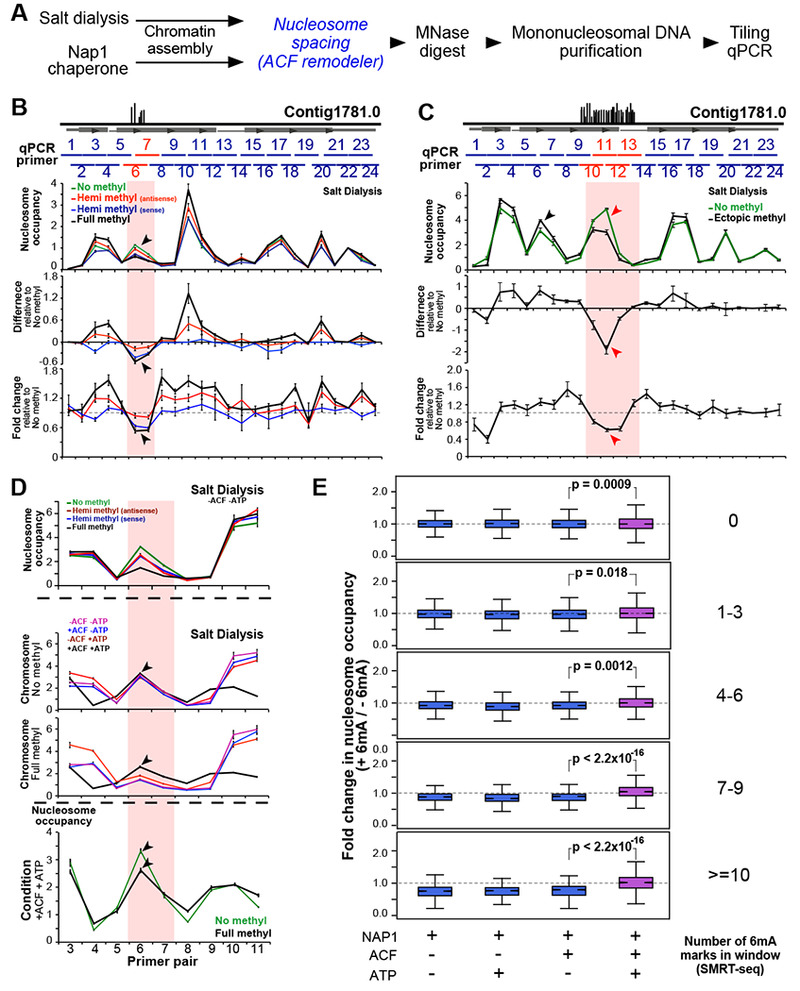
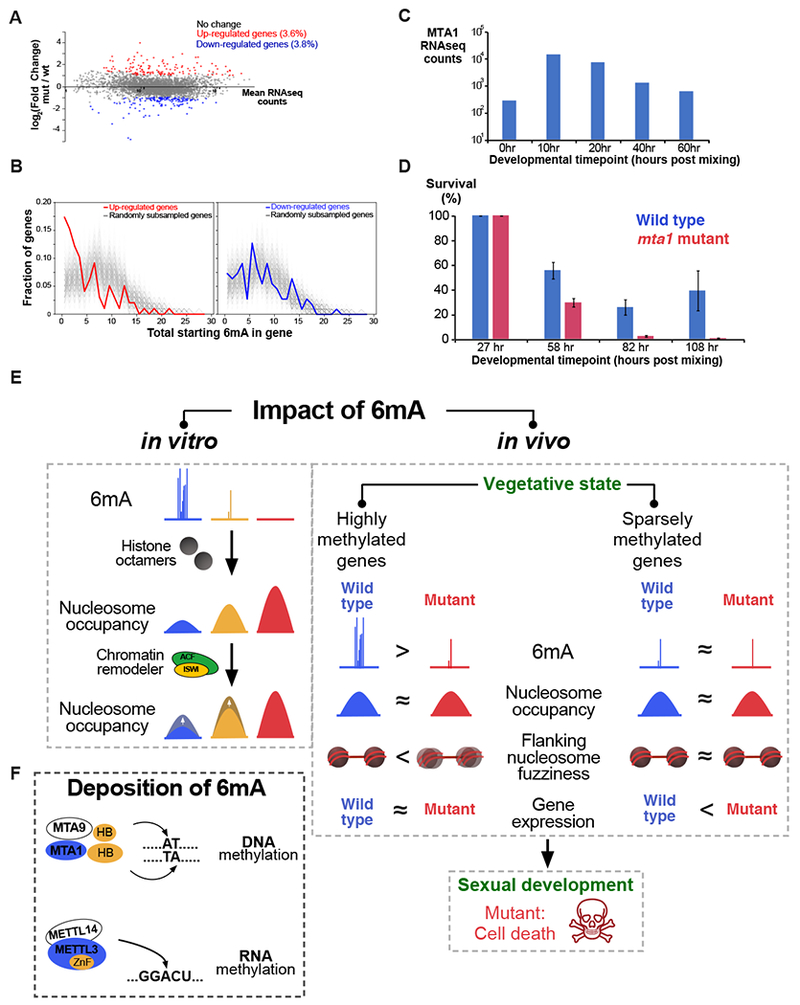
DNA N6-adenine methylation (6mA) has recently been described in diverse eukaryotes, spanning unicellular organisms to metazoa. Here, we report a DNA 6mA methyltransferase complex in ciliates, termed MTA1c. It consists of two MT-A70 proteins and two homeobox-like DNA-binding proteins and specifically methylates dsDNA. Disruption of the catalytic subunit, MTA1, in the ciliate Oxytricha leads to genome-wide loss of 6mA and abolishment of the consensus ApT dimethylated motif. Mutants fail to complete the sexual cycle, which normally coincides with peak MTA1 expression. We investigate the impact of 6mA on nucleosome occupancy in vitro by reconstructing complete, full-length Oxytricha chromosomes harboring 6mA in native or ectopic positions. We show that 6mA directly disfavors nucleosomes in vitro in a local, quantitative manner, independent of DNA sequence. Furthermore, the chromatin remodeler ACF can overcome this effect. Our study identifies a diverged DNA N6-adenine methyltransferase and defines the role of 6mA in chromatin organization.
Keywords: DNA methylation; N6-methyladenine; chromatin remodeling; nucleosome positioning; synthetic chromosomes.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
