

Agglutinin-Like Sequence (ALS) Genes in the Candida parapsilosis Species Complex: Blurring the Boundaries Between Gene Families That Encode Cell-Wall Proteins
- PMID: 31105652
- PMCID: PMC6499006
- DOI: 10.3389/fmicb.2019.00781
Agglutinin-Like Sequence (ALS) Genes in the Candida parapsilosis Species Complex: Blurring the Boundaries Between Gene Families That Encode Cell-Wall Proteins
Abstract
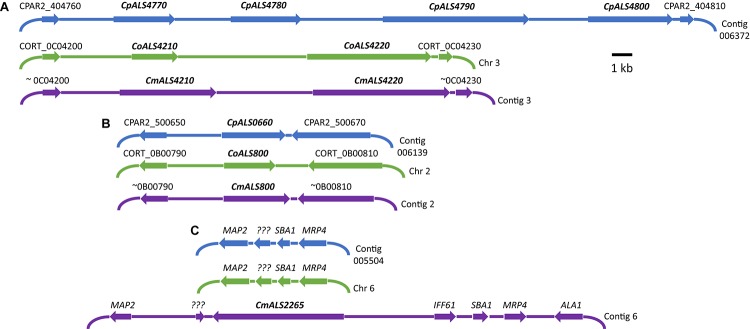
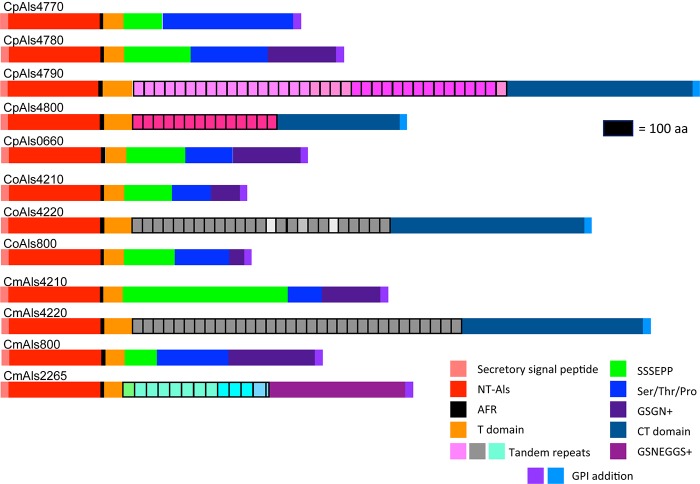
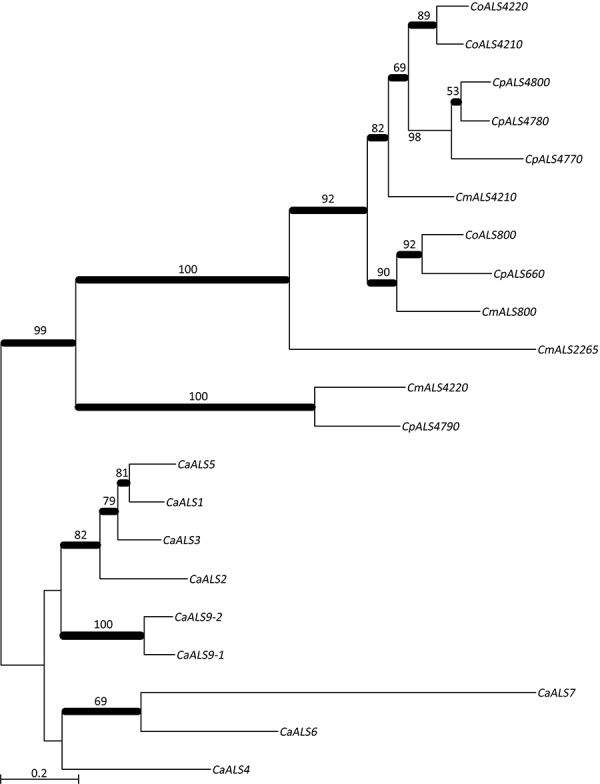
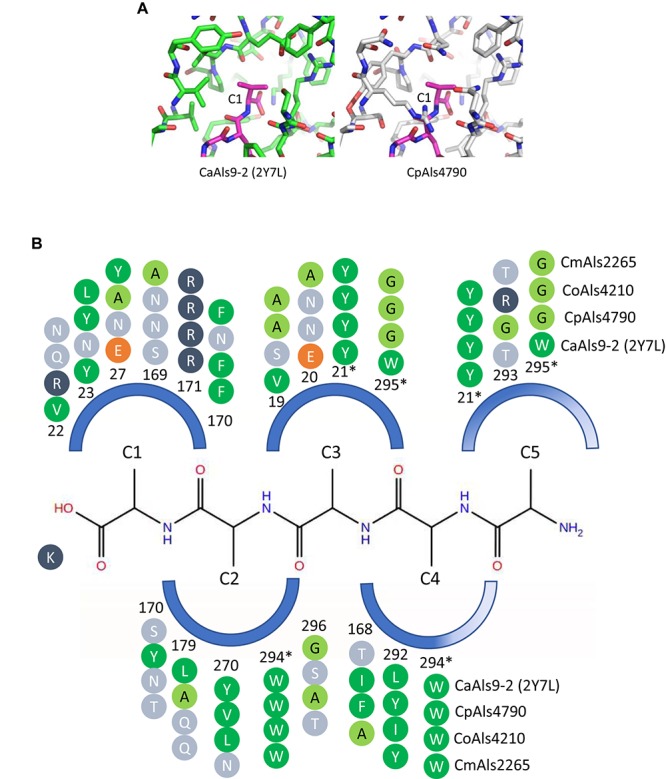
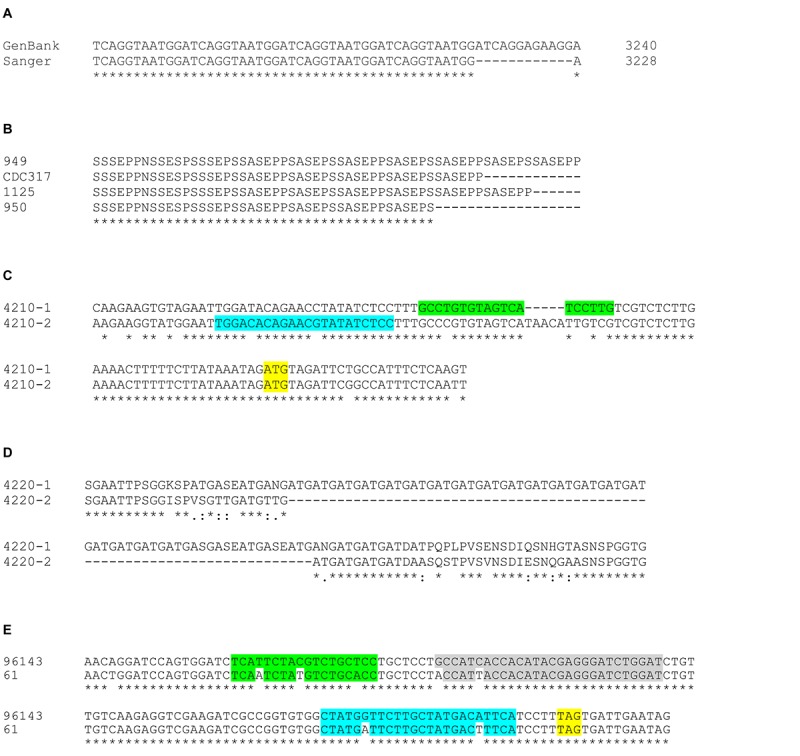
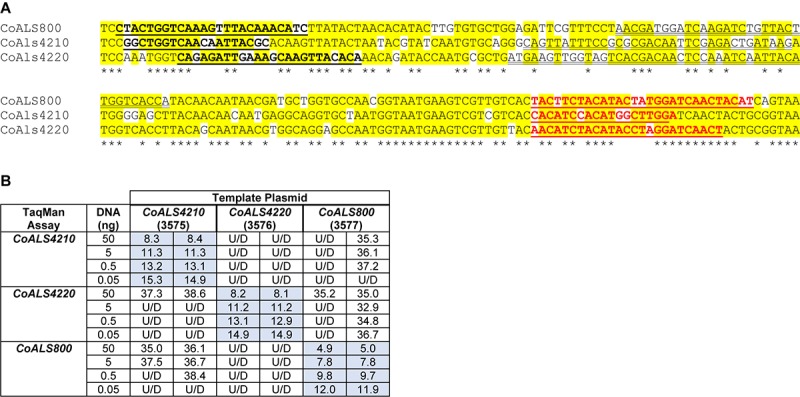
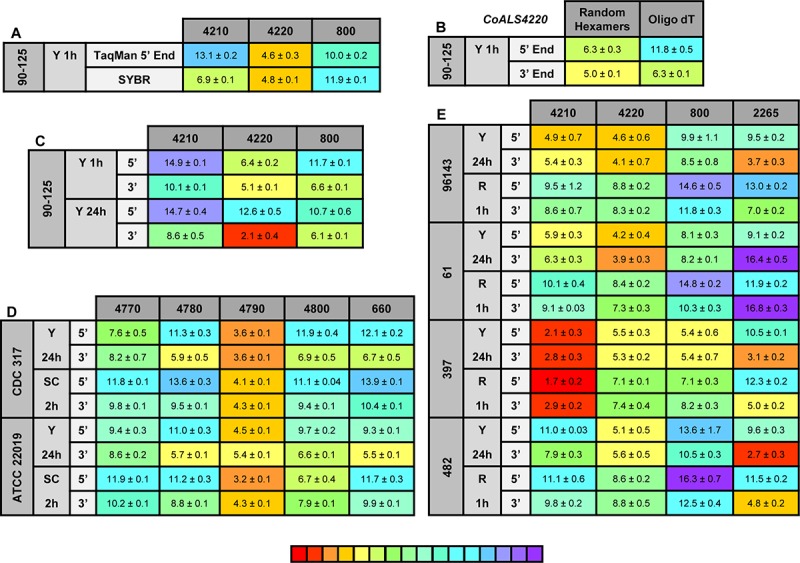
The agglutinin-like sequence (Als) proteins are best-characterized in Candida albicans and known for their role in adhesion of the fungal cell to host and abiotic surfaces. ALS sequences are often misassembled in whole-genome sequence data because each species has multiple ALS loci that contain similar sequences, most notably tandem copies of highly conserved repeated sequences. The Candida parapsilosis species complex includes Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis, three distinct but closely related species. Using publicly available genome resources, de novo genome assemblies, and laboratory experimentation including Sanger sequencing, five ALS genes were characterized in C. parapsilosis strain CDC317, three in C. orthopsilosis strain 90-125, and four in C. metapsilosis strain ATCC 96143. The newly characterized ALS genes shared similar features with the well-known C. albicans ALS family, but also displayed unique attributes such as novel short, imperfect repeat sequences that were found in other genes encoding fungal cell-wall proteins. Evidence of recombination between ALS sequences and other genes was most obvious in CmALS2265, which had the 5' end of an ALS gene and the repeated sequences and 3' end from the IFF/HYR family. Together, these results blur the boundaries between the fungal cell-wall families that were defined in C. albicans. TaqMan assays were used to quantify relative expression for each ALS gene. Some measurements were complicated by the assay location within the ALS gene. Considerable variation was noted in relative gene expression for isolates of the same species. Overall, however, there was a trend toward higher relative gene expression in saturated cultures rather than younger cultures. This work provides a complete description of the ALS genes in the C. parapsilosis species complex and a toolkit that promotes further investigations into the role of the Als proteins in host-fungal interactions.
Keywords: ALS family; Candida species; adhesion; agglutinin-like sequence genes; cell-wall proteins; fungi.
Figures

Similar articles
-
Characterization of the Candida orthopsilosis agglutinin-like sequence (ALS) genes.PLoS One. 2019 Apr 24;14(4):e0215912. doi: 10.1371/journal.pone.0215912. eCollection 2019. PLoS One. 2019. PMID: 31017950 Free PMC article.
-
Pursuing Advances in DNA Sequencing Technology to Solve a Complex Genomic Jigsaw Puzzle: The Agglutinin-Like Sequence (ALS) Genes of Candida tropicalis.Front Microbiol. 2021 Jan 20;11:594531. doi: 10.3389/fmicb.2020.594531. eCollection 2020. Front Microbiol. 2021. PMID: 33552012 Free PMC article.
-
Sequence and analysis of the genome of the pathogenic yeast Candida orthopsilosis.PLoS One. 2012;7(4):e35750. doi: 10.1371/journal.pone.0035750. Epub 2012 Apr 26. PLoS One. 2012. PMID: 22563396 Free PMC article.
-
Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family--a sticky pursuit.Med Mycol. 2008 Feb;46(1):1-15. doi: 10.1080/13693780701435317. Med Mycol. 2008. PMID: 17852717 Free PMC article. Review.
-
The ALS gene family of Candida albicans.Trends Microbiol. 2001 Apr;9(4):176-80. doi: 10.1016/s0966-842x(01)01984-9. Trends Microbiol. 2001. PMID: 11286882 Review.
Cited by
-
Candida albicans evades NK cell elimination via binding of Agglutinin-Like Sequence proteins to the checkpoint receptor TIGIT.Nat Commun. 2022 May 5;13(1):2463. doi: 10.1038/s41467-022-30087-z. Nat Commun. 2022. PMID: 35513379 Free PMC article.
-
The Flo Adhesin Family.Pathogens. 2021 Oct 28;10(11):1397. doi: 10.3390/pathogens10111397. Pathogens. 2021. PMID: 34832553 Free PMC article. Review.
-
Candida Infections: The Role of Saliva in Oral Health-A Narrative Review.Microorganisms. 2025 Mar 23;13(4):717. doi: 10.3390/microorganisms13040717. Microorganisms. 2025. PMID: 40284554 Free PMC article. Review.
-
Genetic Manipulation as a Tool to Unravel Candida parapsilosis Species Complex Virulence and Drug Resistance: State of the Art.J Fungi (Basel). 2021 Jun 7;7(6):459. doi: 10.3390/jof7060459. J Fungi (Basel). 2021. PMID: 34200514 Free PMC article. Review.
-
Identification of a novel Candida metapsilosis isolate reveals multiple hybridization events.G3 (Bethesda). 2022 Jan 4;12(1):jkab367. doi: 10.1093/g3journal/jkab367. G3 (Bethesda). 2022. PMID: 34791169 Free PMC article.
References
-
- Applied Biosystems Inc. (2008). Guide to Performing Relative Quantitation of Gene Expression using Real-Time Quantitative PCR. Available at: https://www.thermofisher.com/document-connect/document-connect.html?url=... (accessed December 20, 2017).
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous