

HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells
- PMID: 31105999
- PMCID: PMC6511643
HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells
Abstract
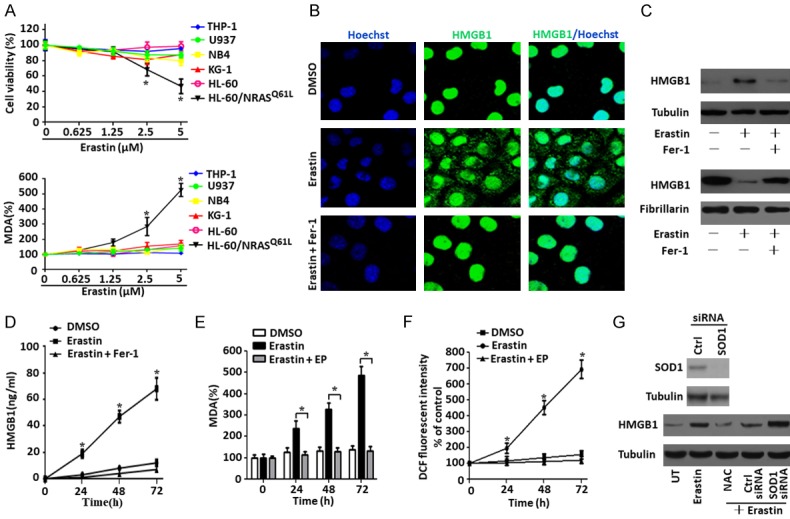
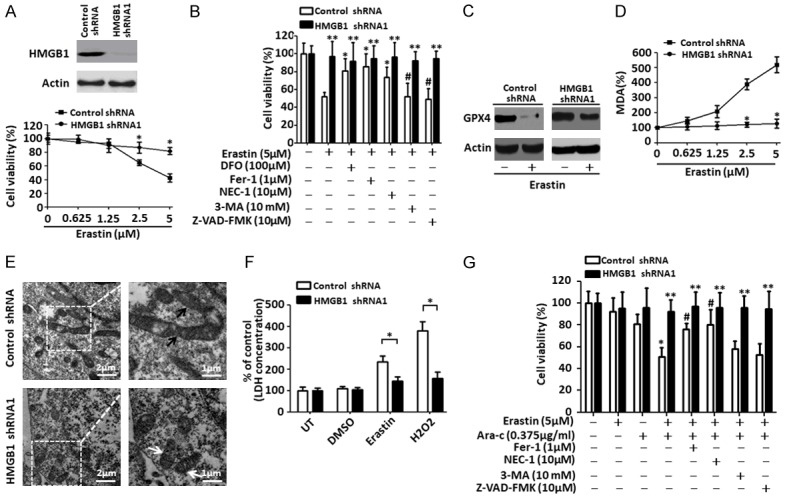
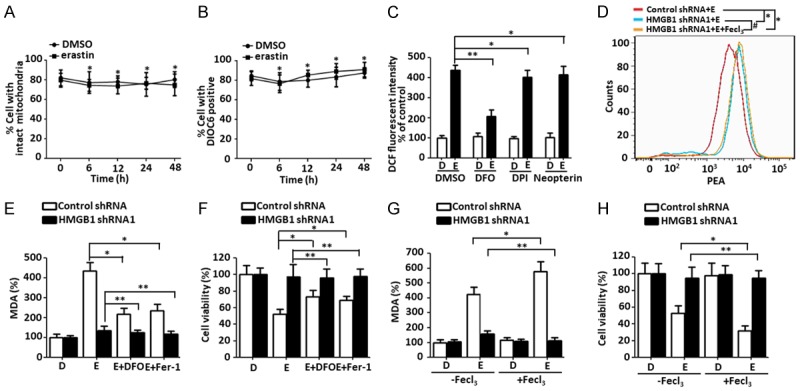
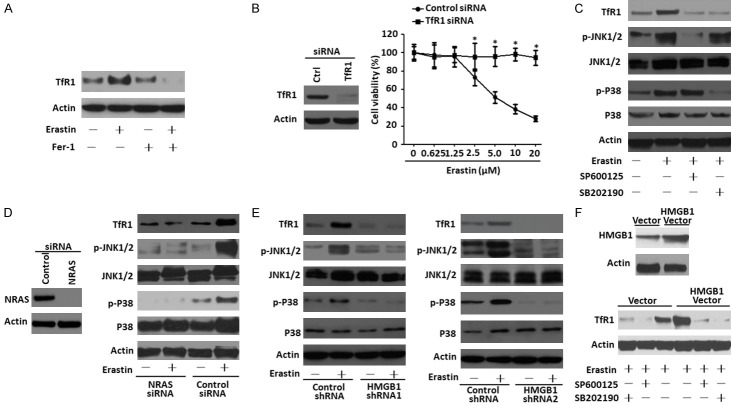
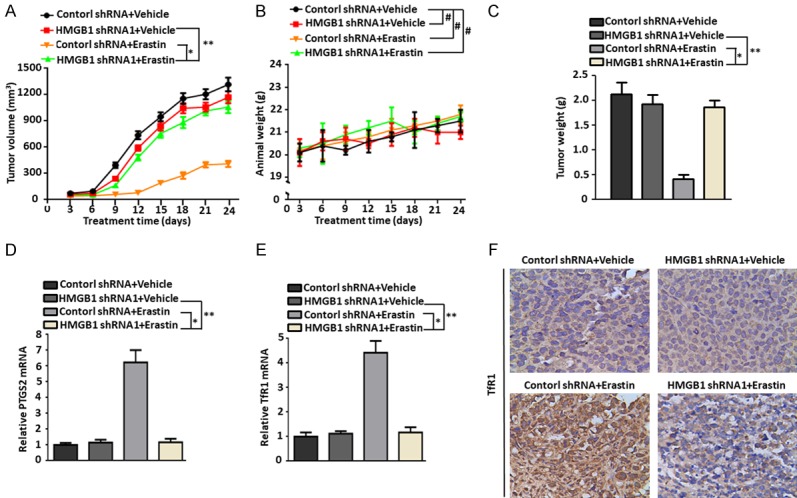
Ferroptosis is emerging as a new form of regulated cell death driven by oxidative injury promoting lipid peroxidation in an iron-dependent manner. High mobility group box 1 (HMGB1) plays an important role in leukemia pathogenesis and chemotherapy resistance. The mechanisms of ferroptosis in tumor pathogenesis and treatment have been a recent research focus but the role of HMGB1 in regulating ferroptosis especially in leukemia still remains largely unknown. Here, we shown that HMGB1 is a critical regulator of eratin-induced ferroptosis in HL-60 cell line expressing NRASQ61L (HL-60/NRASQ61L). Erastin enhanced ROS levels, thereby promoting cytosolic translocation of HMGB1 and enhancing cell death. Knockdown of HMGB1 decreased erastin-induced ROS generation and cell death in an iron-mediated lysosomal pathway in HL-60/NRASQ61L cells. Knockdown of HMGB1 or rat sarcoma (RAS), or pharmacological inhibition of JNK and p38 decreased TfR1 levels in HL-60/NRASQ61L cells. Importantly, these data were further supported by our in vivo experiment, in which xenografts formed by HMGB1 knockdown HL-60/NRASQ61L cells had lower PTGS2 and TfR1 expression than that in control mice. Taken together, these results suggest that HMGB1 is a novel regulator of ferroptosis via the RAS-JNK/p38 pathway and a potential drug target for therapeutic interventions in leukemia.
Keywords: HMGB1; MAPK; acute myeloid leukemia; ferroptosis; transferrin receptor 1.
Conflict of interest statement
None.
Figures
Similar articles
-
The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents.Mol Cell Oncol. 2015 May 26;2(4):e1054549. doi: 10.1080/23723556.2015.1054549. eCollection 2015 Oct-Dec. Mol Cell Oncol. 2015. PMID: 27308510 Free PMC article.
-
Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells.Eur Rev Med Pharmacol Sci. 2018 Jun;22(12):3826-3836. doi: 10.26355/eurrev_201806_15267. Eur Rev Med Pharmacol Sci. 2018. PMID: 29949159
-
HMGB1 regulates ferroptosis through Nrf2 pathway in mesangial cells in response to high glucose.Biosci Rep. 2021 Feb 26;41(2):BSR20202924. doi: 10.1042/BSR20202924. Biosci Rep. 2021. PMID: 33565572 Free PMC article.
-
The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy.Onco Targets Ther. 2020 Jun 11;13:5429-5441. doi: 10.2147/OTT.S254995. eCollection 2020. Onco Targets Ther. 2020. PMID: 32606760 Free PMC article. Review.
-
Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis.Curr Top Microbiol Immunol. 2017;403:143-170. doi: 10.1007/82_2016_508. Curr Top Microbiol Immunol. 2017. PMID: 28204974 Review.
Cited by
-
Is ferroptosis involved in ROS-induced testicular lesions in a varicocele rat model?Basic Clin Androl. 2021 Apr 1;31(1):10. doi: 10.1186/s12610-021-00125-9. Basic Clin Androl. 2021. PMID: 33789564 Free PMC article.
-
20-HETE Participates in Intracerebral Hemorrhage-Induced Acute Injury by Promoting Cell Ferroptosis.Front Neurol. 2021 Nov 12;12:763419. doi: 10.3389/fneur.2021.763419. eCollection 2021. Front Neurol. 2021. PMID: 34867747 Free PMC article.
-
Ferroptosis as a mechanism of non-ferrous metal toxicity.Arch Toxicol. 2022 Sep;96(9):2391-2417. doi: 10.1007/s00204-022-03317-y. Epub 2022 Jun 21. Arch Toxicol. 2022. PMID: 35727353 Review.
-
Induction of ferroptosis by SIRT1 knockdown alleviates cytarabine resistance in acute myeloid leukemia by activating the HMGB1/ACSL4 pathway.Int J Oncol. 2025 Jan;66(1):2. doi: 10.3892/ijo.2024.5708. Epub 2024 Nov 22. Int J Oncol. 2025. PMID: 39575456 Free PMC article.
-
Immunogenic cell death in cancer immunotherapy.BMB Rep. 2023 May;56(5):275-286. doi: 10.5483/BMBRep.2023-0024. BMB Rep. 2023. PMID: 37081756 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous