

Heart Inflammation: Immune Cell Roles and Roads to the Heart
- PMID: 31108102
- PMCID: PMC6717912
- DOI: 10.1016/j.ajpath.2019.04.009
Heart Inflammation: Immune Cell Roles and Roads to the Heart
Abstract
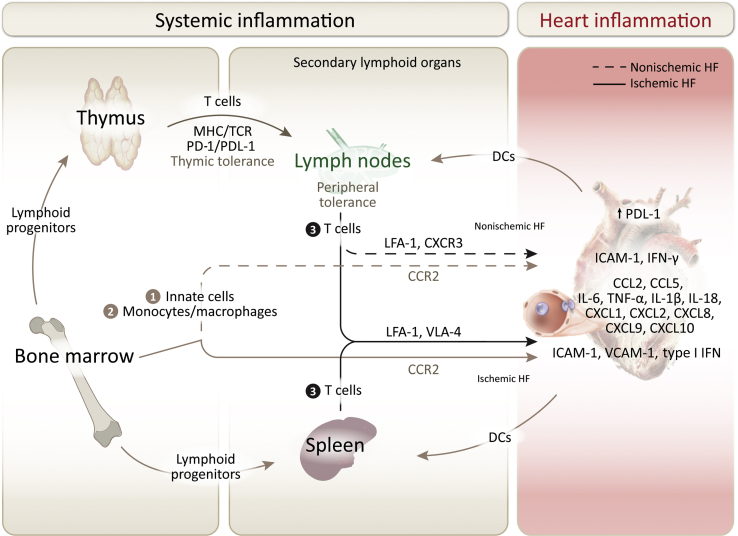
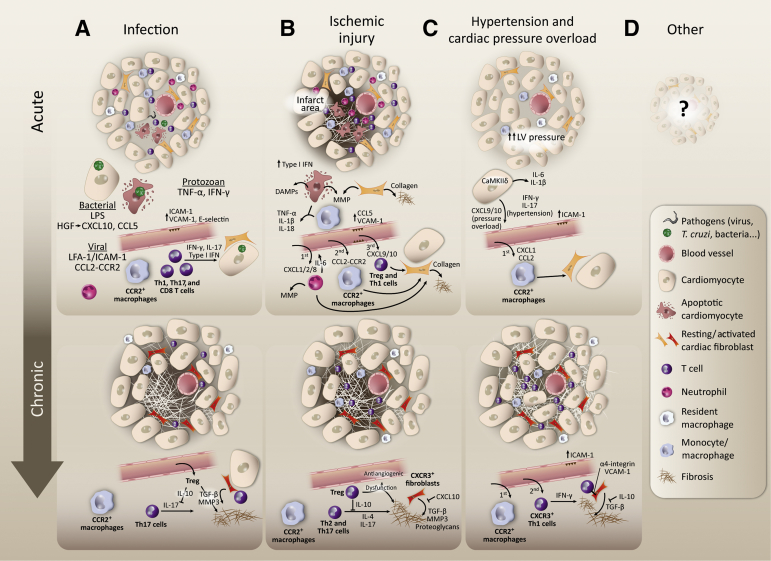
Heart failure (HF) has been traditionally viewed as a disease of the cardiac muscle associated with systemic inflammation. Burgeoning evidence implicates immune effector mechanisms that include immune cell activation and trafficking to the heart. Immune cell infiltration in the myocardium can have adverse effects in the heart and contribute to the pathogenesis of HF. Both innate and adaptive immunity operate sequentially, and the specificity of these responses depends on the initial trigger sensed by the heart. Although the role of the immune system in the initial inflammatory response to infection and injury is well studied, what sets the trajectory to HF from different etiologies and the role of immunity once HF has been established is less understood. Herein, we review experimental and clinical knowledge of cardiac inflammation induced by different triggers that often result in HF from different etiologies. We focus on the mechanisms of immune cell activation systemically and on the pathways immune cells use to traffic to the heart.
Copyright © 2019 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Role of Adaptive Immunity in the Development and Progression of Heart Failure: New Evidence.Arch Med Res. 2017 Jan;48(1):1-11. doi: 10.1016/j.arcmed.2016.12.008. Arch Med Res. 2017. PMID: 28577862 Review.
-
A novel 72-kDa leukocyte-derived osteoglycin enhances the activation of toll-like receptor 4 and exacerbates cardiac inflammation during viral myocarditis.Cell Mol Life Sci. 2017 Apr;74(8):1511-1525. doi: 10.1007/s00018-016-2423-7. Epub 2016 Nov 23. Cell Mol Life Sci. 2017. PMID: 27878326 Free PMC article.
-
Protecting the pump: controlling myocardial inflammatory responses.Annu Rev Physiol. 2006;68:67-95. doi: 10.1146/annurev.physiol.68.040104.124611. Annu Rev Physiol. 2006. PMID: 16460267 Review.
-
Role of the innate immune system in acute viral myocarditis.Basic Res Cardiol. 2009 May;104(3):228-37. doi: 10.1007/s00395-008-0765-5. Epub 2009 Jan 21. Basic Res Cardiol. 2009. PMID: 19159057 Free PMC article.
-
Immune cell infiltration landscapes in pediatric acute myocarditis analyzed by CIBERSORT.J Cardiol. 2021 Feb;77(2):174-178. doi: 10.1016/j.jjcc.2020.08.004. Epub 2020 Sep 3. J Cardiol. 2021. PMID: 32891480
Cited by
-
Nicotinamide Riboside Alleviates Cardiac Dysfunction and Remodeling in Pressure Overload Cardiac Hypertrophy.Oxid Med Cell Longev. 2021 Sep 15;2021:5546867. doi: 10.1155/2021/5546867. eCollection 2021. Oxid Med Cell Longev. 2021. PMID: 34567409 Free PMC article.
-
The Gut-Heart Axis and Its Role in Doxorubicin-Induced Cardiotoxicity: A Narrative Review.Microorganisms. 2025 Apr 9;13(4):855. doi: 10.3390/microorganisms13040855. Microorganisms. 2025. PMID: 40284691 Free PMC article. Review.
-
Heart failure decompensation with cardiogenic shock exhibits distinct sequential inflammatory profiles.ESC Heart Fail. 2025 Jun;12(3):2077-2086. doi: 10.1002/ehf2.15217. Epub 2025 Feb 9. ESC Heart Fail. 2025. PMID: 39925014 Free PMC article.
-
Understanding the Mechanism of Cardiotoxicity Induced by Nanomaterials: A Comprehensive Review.Small Sci. 2025 Feb 20;5(5):2400498. doi: 10.1002/smsc.202400498. eCollection 2025 May. Small Sci. 2025. PMID: 40395348 Free PMC article. Review.
-
Bidirectional Crosstalk between the Heart and Brain in Alzheimer's Disease.Aging Dis. 2024 Oct 31;16(5):2979-2998. doi: 10.14336/AD.2024.1132. Aging Dis. 2024. PMID: 39571156 Free PMC article. Review.
References
-
- Benjamin E.J., Virani S.S., Callaway C.W., Chamberlain A.M., Chang A.R., Cheng S., American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics-2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. - PubMed
-
- Elster S.K., Braunwald E., Wood H.F. A study of C-reactive protein in the serum of patients with congestive heart failure. Am Heart J. 1956;51:533–541. - PubMed
-
- Chung E.S., Packer M., Lo K.H., Fasanmade A.A., Willerson J.T., Anti-TNF Therapy Against Congestive Heart Failure Investigators Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the Anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107:3133–3140. - PubMed
-
- Mann D.L., McMurray J.J., Packer M., Swedberg K., Borer J.S., Colucci W.S., Djian J., Drexler H., Feldman A., Kober L., Krum H., Liu P., Nieminen M., Tavazzi L., van Veldhuisen D.J., Waldenstrom A., Warren M., Westheim A., Zannad F., Fleming T. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004;109:1594–1602. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
