

Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor
- PMID: 31108174
- PMCID: PMC6996243
- DOI: 10.1016/j.nbd.2019.05.009
Pridopidine protects neurons from mutant-huntingtin toxicity via the sigma-1 receptor
Abstract
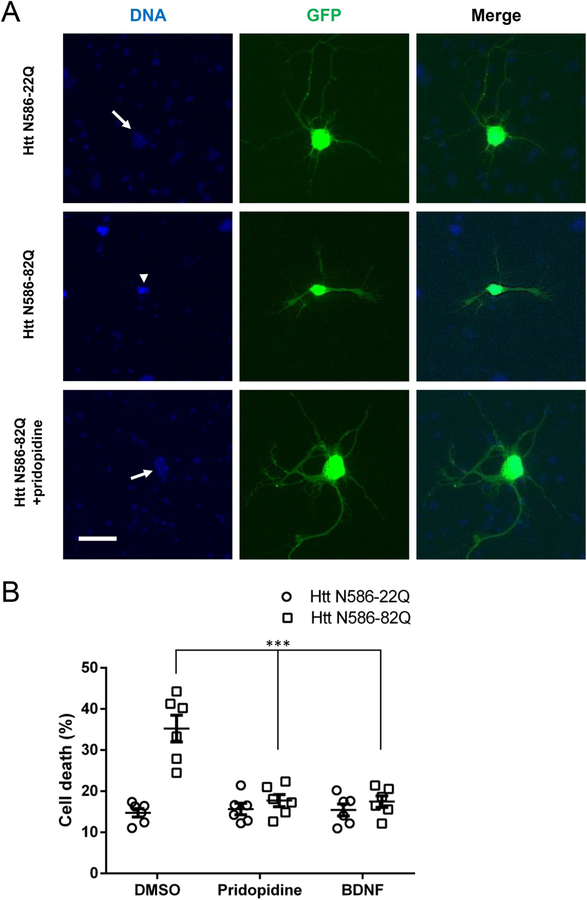
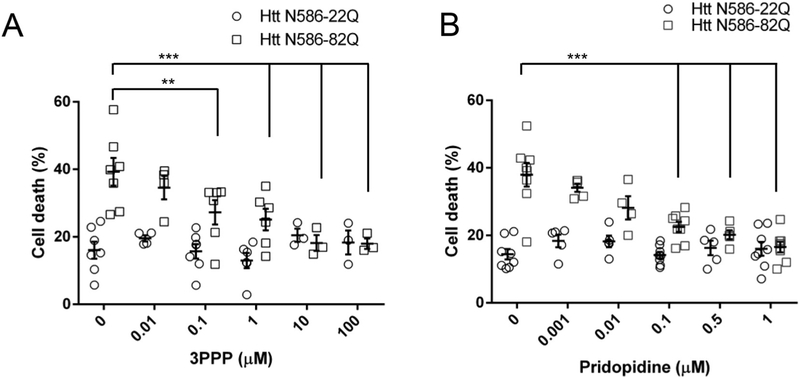
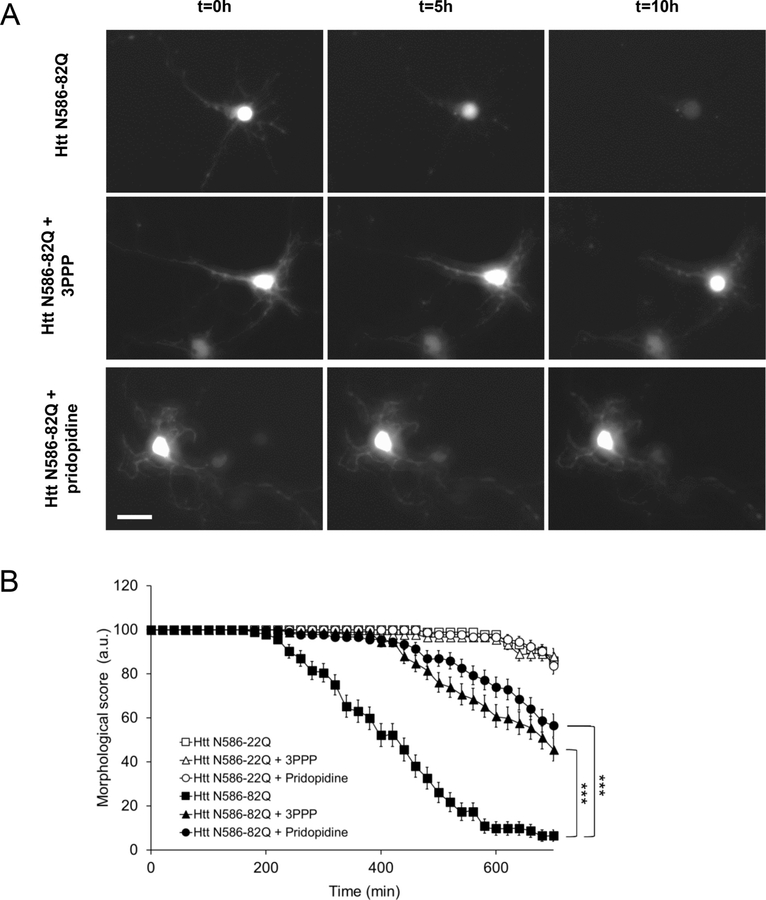
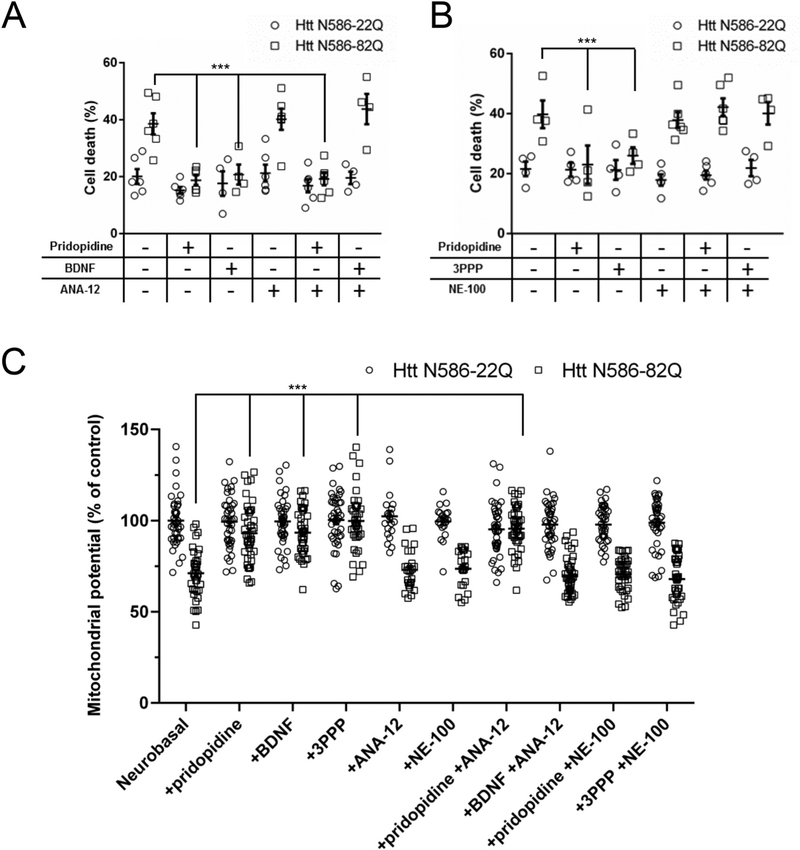
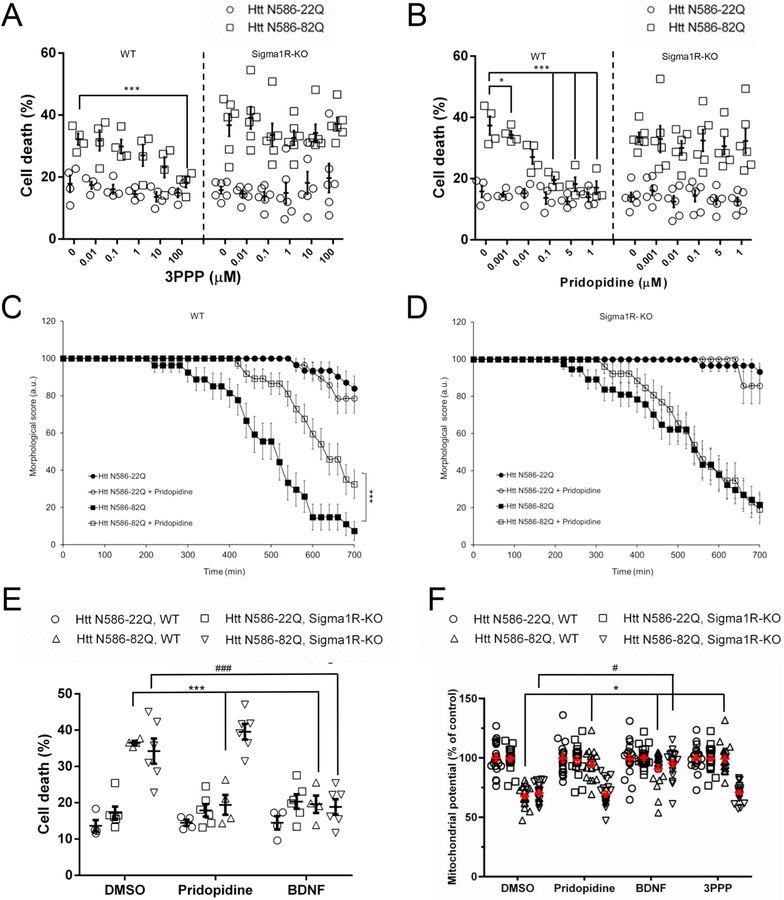
Huntington's disease (HD) is a neurodegenerative disease caused by a CAG repeat expansion in the Huntingtin gene (HTT), translated into a Huntingtin protein with a polyglutamine expansion. There is preferential loss of medium spiny neurons within the striatum and cortical pyramidal neurons. Pridopidine is a small molecule showing therapeutic potential in HD preclinical and clinical studies. Pridopidine has nanomolar affinity to the sigma-1 receptor (sigma-1R), which is located predominantly at the endoplasmic reticulum (ER) and mitochondrial associated ER membrane, and activates neuroprotective pathways. Here we evaluate the neuroprotective effects of pridopidine against mutant Huntingtin toxicity in mouse and human derived in vitro cell models. We also investigate the involvement of the sigma-1 receptor in the mechanism of pridopidine. Pridopidine protects mutant Huntingtin transfected mouse primary striatal and cortical neurons, with an EC50 in the mid nanomolar range, as well as HD patient-derived induced pluripotent stem cells (iPSCs). This protection by pridopidine is blocked by NE-100, a purported sigma-1 receptor antagonist, and not blocked by ANA-12, a reported TrkB receptor antagonist. 3PPP, a documented sigma-1 receptor agonist, shows similar neuroprotective effects. Genetic knock out of the sigma-1 receptor dramatically decreases protection from pridopidine and 3PPP, but not protection via brain derived neurotrophic factor (BDNF). The neuroprotection afforded by pridopidine in our HD cell models is robust and sigma-1 receptor dependent. These studies support the further development of pridopidine, and other sigma-1 receptor agonists as neuroprotective agents for HD and perhaps for other disorders.
Keywords: 3PPP; Huntingtin toxicity; Huntington's disease; Mutant-huntingtin; Neuroprotection; Patient-derived induced pluripotent stem cells; Pridopidine; Primary neurons; Sigma-1 receptor.
Copyright © 2019. Published by Elsevier Inc.
Figures
Similar articles
-
The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease.Neurobiol Dis. 2017 Jan;97(Pt A):46-59. doi: 10.1016/j.nbd.2016.10.006. Epub 2016 Nov 3. Neurobiol Dis. 2017. PMID: 27818324 Free PMC article.
-
Pridopidine rescues BDNF/TrkB trafficking dynamics and synapse homeostasis in a Huntington disease brain-on-a-chip model.Neurobiol Dis. 2022 Oct 15;173:105857. doi: 10.1016/j.nbd.2022.105857. Epub 2022 Sep 6. Neurobiol Dis. 2022. PMID: 36075537
-
Large-scale transcriptomic analysis reveals that pridopidine reverses aberrant gene expression and activates neuroprotective pathways in the YAC128 HD mouse.Mol Neurodegener. 2018 May 21;13(1):25. doi: 10.1186/s13024-018-0259-3. Mol Neurodegener. 2018. PMID: 29783994 Free PMC article.
-
Pridopidine in the treatment of Huntington's disease.Rev Neurosci. 2020 May 26;31(4):441-451. doi: 10.1515/revneuro-2019-0085. Rev Neurosci. 2020. PMID: 32083454 Review.
-
Normalization of Calcium Balance in Striatal Neurons in Huntington's Disease: Sigma 1 Receptor as a Potential Target for Therapy.Biochemistry (Mosc). 2021 Apr;86(4):471-479. doi: 10.1134/S0006297921040076. Biochemistry (Mosc). 2021. PMID: 33941067 Review.
Cited by
-
Mutational Analysis of Sigma-1 Receptor's Role in Synaptic Stability.Front Neurosci. 2019 Sep 19;13:1012. doi: 10.3389/fnins.2019.01012. eCollection 2019. Front Neurosci. 2019. PMID: 31607852 Free PMC article.
-
Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases?Antioxidants (Basel). 2022 Jan 15;11(1):165. doi: 10.3390/antiox11010165. Antioxidants (Basel). 2022. PMID: 35052668 Free PMC article. Review.
-
PB28, the Sigma-1 and Sigma-2 Receptors Modulator With Potent Anti-SARS-CoV-2 Activity: A Review About Its Pharmacological Properties and Structure Affinity Relationships.Front Pharmacol. 2020 Dec 7;11:589810. doi: 10.3389/fphar.2020.589810. eCollection 2020. Front Pharmacol. 2020. PMID: 33364961 Free PMC article. Review.
-
Transmembrane Protein 230 Mediates a Poly(ADP-ribose) Polymerase-1-Linked Apoptosis.Front Aging Neurosci. 2020 Aug 7;12:235. doi: 10.3389/fnagi.2020.00235. eCollection 2020. Front Aging Neurosci. 2020. PMID: 32848711 Free PMC article.
-
The impact of cannabinoid type 2 receptors (CB2Rs) in neuroprotection against neurological disorders.Acta Pharmacol Sin. 2020 Dec;41(12):1507-1518. doi: 10.1038/s41401-020-00530-2. Epub 2020 Oct 6. Acta Pharmacol Sin. 2020. PMID: 33024239 Free PMC article. Review.
References
-
- Albayrak Y, Hashimoto K, 2017. Sigma-1 receptor agonists and their clinical implications in neuropsychiatric disorders. Adv. Exp. Med. Biol 964, 153–161. - PubMed
-
- Allahtavakoli M, Jarrott B, 2011. Sigma-1 receptor ligand PRE-084 reduced infarct volume, neurological deficits, pro-inflammatory cytokines and enhanced anti-inflammatory cytokines after embolic stroke in rats. Brain Res. Bull 85, 219–224. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
