

A patient with glycogen storage disease type Ia combined with chronic hepatitis B infection: a case report
- PMID: 31109299
- PMCID: PMC6528214
- DOI: 10.1186/s12881-019-0816-9
A patient with glycogen storage disease type Ia combined with chronic hepatitis B infection: a case report
Abstract
Background: Glycogen storage disease type I (GSD I), also known as von Gierk disease, is a metabolic disorder leading to the excessive accumulation of glycogen and fat in organs, characterized by hepatomegaly, hypoglycemia, lactic acidemia, hyperlipidemia, hyperuricemia, puberty delay and growth retardation, which can be indicated by height, weight, blood glucose and blood lipids.
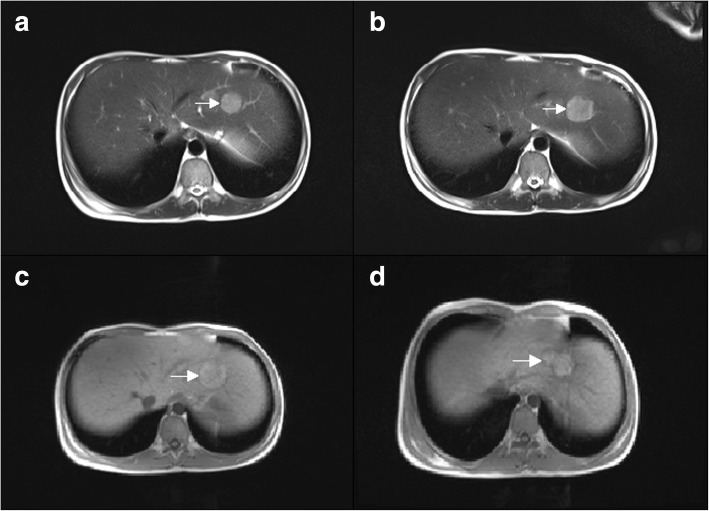
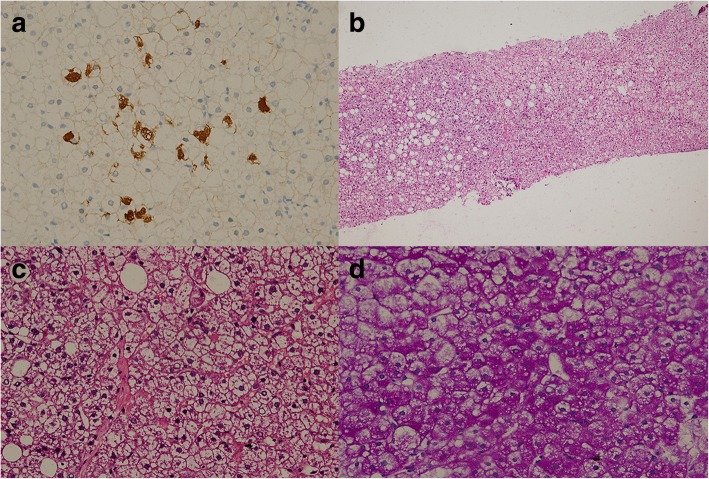
Case presentation: Here we present a 16-year-old male patient with GSD Ia complicated with hepatic adenoma and combined with hepatitis B. As a chronic hepatitis B patient, the patient was admitted to hospital in order to further clarify the nature of hepatic space occupancy because of suspicion of hepatocellular carcinoma. However, the imaging studies did not support hepatocellular carcinoma certainly. And by tracing his clinical history, we suggested that he might suffer from GSD I. Finally the diagnosis was confirmed by MRI (Gd-EOB-DTPA), liver biopsy and whole exome sequencing (WES). The WES discovered a homozygous point mutation at the exon 5 of G6PC gene at 17th chromosome, c.G648 T (p.L216 L, NM_000151, rs80356484). This pathogenic mutation causes CTG changing to CTT at protein 216. Though both codons encode leucine, this silent mutation creates a new splicing site 91 bp downstream of the authentic splice site. According to previous research, this mutation is a disease causal variant for GSD Ia, and has a high frequency among GSD patients in China and Japan. This patient was finally diagnosed as GSD Ia complicated with hepatic adenoma and combined with chronic hepatitis B, and received corn starch therapy immediately after GSD was suspected. After receiving corn starch therapy, the height and weight of the patient were increased, and the secondary sexual characteristics were developed, including beard, pubic hair and seminal emission. Unexpectedly, the liver adenomas were still increasing, and we did not find any cause to explain this phenomenon.
Conclusion: This patient was diagnosed as GSD Ia combined with chronic hepatitis B, who responded to corn starch intervention. For childhood patients with hypoglycaemia, hyperlipidemia, puberty delay and growth retardation, GSD should be considered. Gene sequencing is valuable for the quick identification of GSD subtypes.
Keywords: Chronic hepatitis B; G6PC gene; GSD Ia; Growth retardation.
Conflict of interest statement
The funding sources had no role in study design, collection, analysis, or interpretation of data, or the writing of the report; or the decision to submit the report for publication.
The authors declare that they have no competing interests.
Figures

Similar articles
-
A glycogen storage disease type 1a patient with type 2 diabetes.BMC Med Genomics. 2022 Sep 27;15(1):205. doi: 10.1186/s12920-022-01344-3. BMC Med Genomics. 2022. PMID: 36167523 Free PMC article.
-
A case study of a liver transplant-treated patient with glycogen storage disease type Ia presenting with multiple inflammatory hepatic adenomas: an analysis of clinicopathologic and genetic data.BMC Med Genomics. 2024 May 6;17(1):124. doi: 10.1186/s12920-024-01888-6. BMC Med Genomics. 2024. PMID: 38711024 Free PMC article.
-
[Heterogeneous phenotypes in Chinese glycogen storage disease type Ia patients with homozygous G727T mutation].Zhonghua Er Ke Za Zhi. 2003 Apr;41(4):252-5. Zhonghua Er Ke Za Zhi. 2003. PMID: 14754525 Chinese.
-
Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease.Hum Mutat. 2008 Jul;29(7):921-30. doi: 10.1002/humu.20772. Hum Mutat. 2008. PMID: 18449899 Free PMC article. Review.
-
Emerging roles of autophagy in hepatic tumorigenesis and therapeutic strategies in glycogen storage disease type Ia: A review.J Inherit Metab Dis. 2021 Jan;44(1):118-128. doi: 10.1002/jimd.12267. Epub 2020 Jul 2. J Inherit Metab Dis. 2021. PMID: 32474930 Review.
Cited by
-
Endocrine involvement in hepatic glycogen storage diseases: pathophysiology and implications for care.Rev Endocr Metab Disord. 2024 Aug;25(4):707-725. doi: 10.1007/s11154-024-09880-2. Epub 2024 Apr 1. Rev Endocr Metab Disord. 2024. PMID: 38556561 Free PMC article. Review.
-
Case Report: Glycogen Storage Disease Type Ia in a Chinese Child Treated With Growth Hormone.Front Pediatr. 2022 Jun 17;10:921323. doi: 10.3389/fped.2022.921323. eCollection 2022. Front Pediatr. 2022. PMID: 35783312 Free PMC article.
-
A glycogen storage disease type 1a patient with type 2 diabetes.BMC Med Genomics. 2022 Sep 27;15(1):205. doi: 10.1186/s12920-022-01344-3. BMC Med Genomics. 2022. PMID: 36167523 Free PMC article.
References
-
- Chen YT. Glycogen storage diseases[M]//Behrman RE. The Nelson textbook of pediatrics. 17th edition. Philadephia, Pennsylvania:WB, Saunders Company,2004:469–475.
-
- Lam CW, But WM, Shek CC, et al. Glucose-6-phosphatase gene (727G–>T) splicing mutation is prevalent in Hong Kong Chinese patients with glycogen storage disease type 1a[J]. Clin Genet. 1998;53:184–90. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
