

Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL
- PMID: 31110043
- PMCID: PMC6664184
- DOI: 10.1074/jbc.RA118.006302
Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL
Abstract
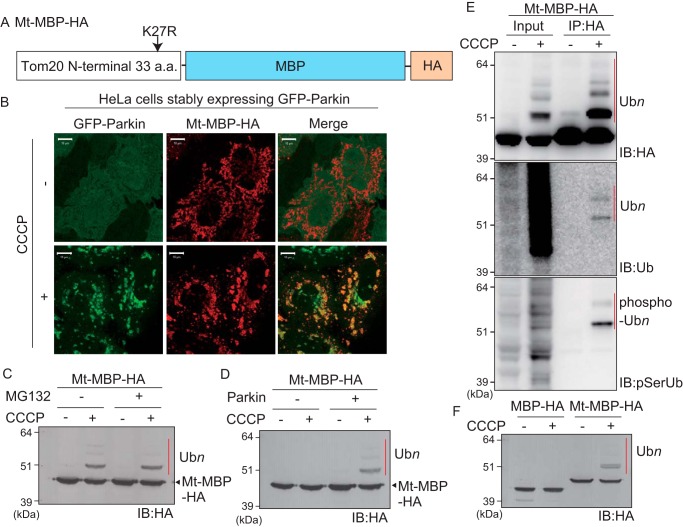
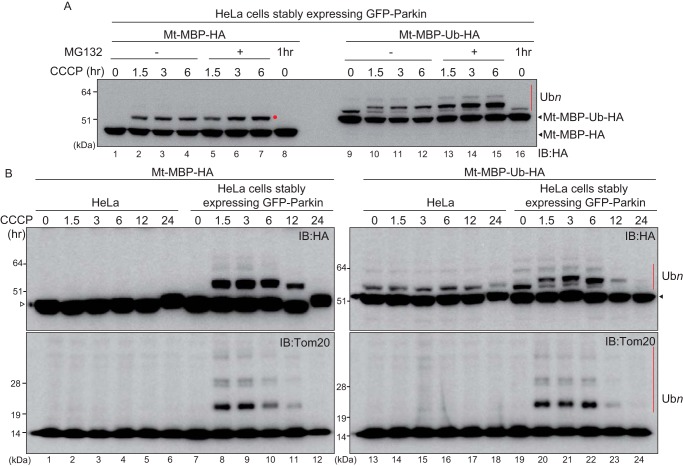
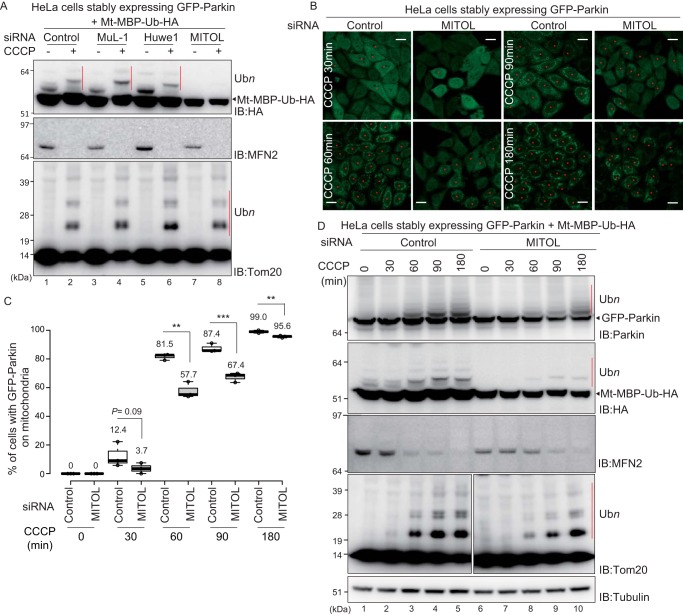
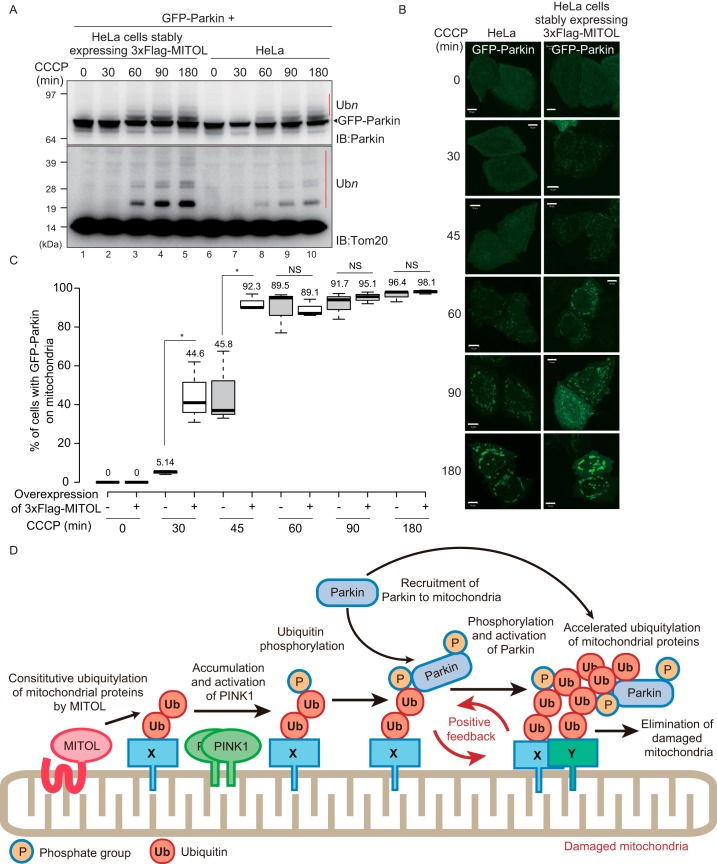
PINK1 (PARK6) and PARKIN (PARK2) are causal genes of recessive familial Parkinson's disease. Parkin is a ubiquitin ligase E3 that conjugates ubiquitin to impaired mitochondrial proteins for organelle degradation. PINK1, a Ser/Thr kinase that accumulates only on impaired mitochondria, phosphorylates two authentic substrates, the ubiquitin-like domain of Parkin and ubiquitin. Our group and others have revealed that both the subcellular localization and ligase activity of Parkin are regulated through interactions with phosphorylated ubiquitin. Once PINK1 localizes on impaired mitochondria, PINK1-catalyzed phosphoubiquitin recruits and activates Parkin. Parkin then supplies a ubiquitin chain to PINK1 for phosphorylation. The amplified ubiquitin functions as a signal for the sequestration and degradation of the damaged mitochondria. Although a bewildering variety of Parkin substrates have been reported, the basis for Parkin substrate specificity remains poorly understood. Moreover, the mechanism underlying initial activation and translocation of Parkin onto mitochondria remains unclear, because the presence of ubiquitin on impaired mitochondria is thought to be a prerequisite for the initial PINK1 phosphorylation process. Here, we show that artificial mitochondria-targeted proteins are ubiquitylated by Parkin, suggesting that substrate specificity of Parkin is not determined by its amino acid sequence. Moreover, recruitment and activation of Parkin are delayed following depletion of the mitochondrial E3, MITOL/March5. We propose a model in which the initial step in Parkin recruitment and activation requires protein ubiquitylation by MITOL/March5 with subsequent PINK1-mediated phosphorylation. Because PINK1 and Parkin amplify the ubiquitin signal via a positive feedback loop, the low substrate specificity of Parkin might facilitate this amplification process.
Keywords: Parkinson disease; mitophagy; parkin; ubiquitin; ubiquitin ligase.
© 2019 Koyano et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures



References
-
- Tanner C. M., Kamel F., Ross G. W., Hoppin J. A., Goldman S. M., Korell M., Marras C., Bhudhikanok G. S., Kasten M., Chade A. R., Comyns K., Richards M. B., Meng C., Priestley B., Fernandez H. H., et al. (2011) Rotenone, paraquat, and Parkinson's disease. Environ. Health Perspect. 119, 866–872 10.1289/ehp.1002839 - DOI - PMC - PubMed
-
- Valente E. M., Abou-Sleiman P. M., Caputo V., Muqit M. M., Harvey K., Gispert S., Ali Z., Del Turco D., Bentivoglio A. R., Healy D. G., Albanese A., Nussbaum R., González-Maldonado R., Deller T., Salvi S., et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160 10.1126/science.1096284 - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
