

Identification of TP53RK-Binding Protein (TPRKB) Dependency in TP53-Deficient Cancers
- PMID: 31110156
- PMCID: PMC6679750
- DOI: 10.1158/1541-7786.MCR-19-0144
Identification of TP53RK-Binding Protein (TPRKB) Dependency in TP53-Deficient Cancers
Abstract
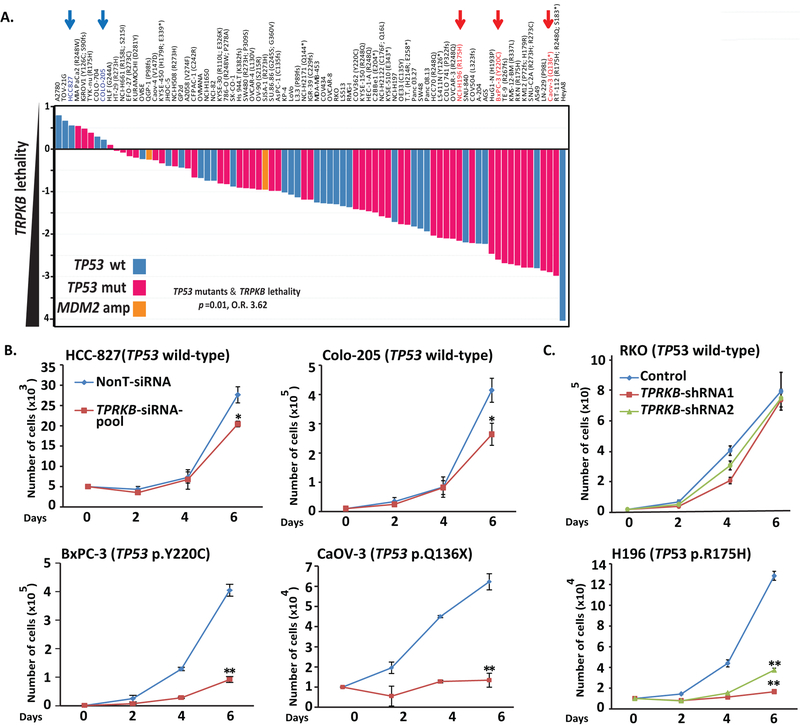
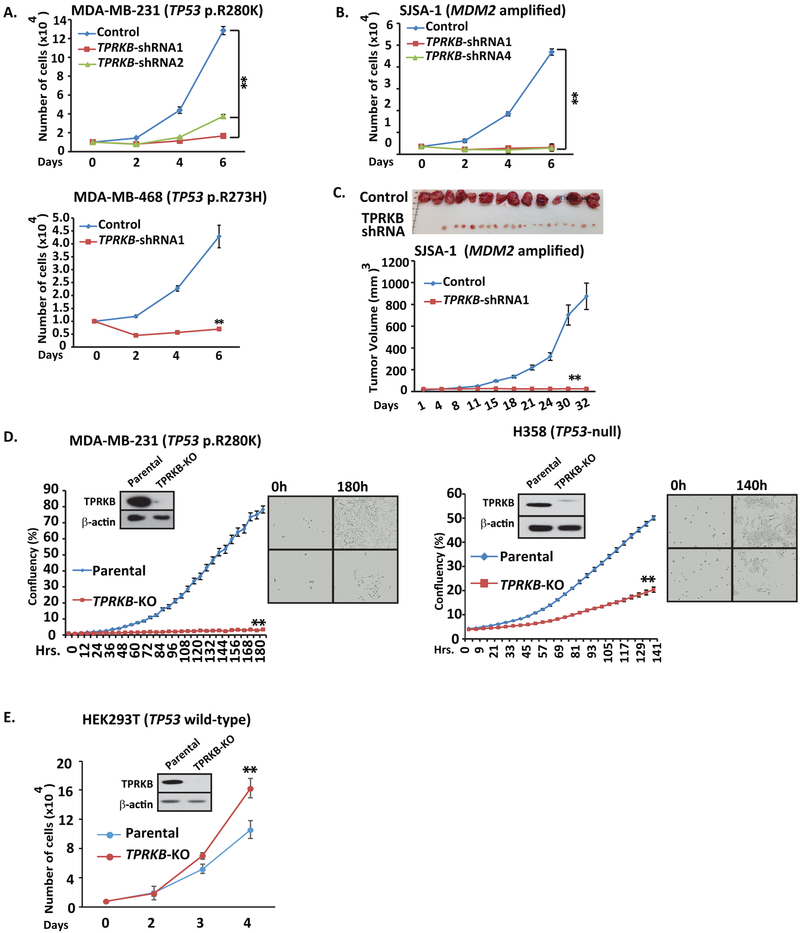
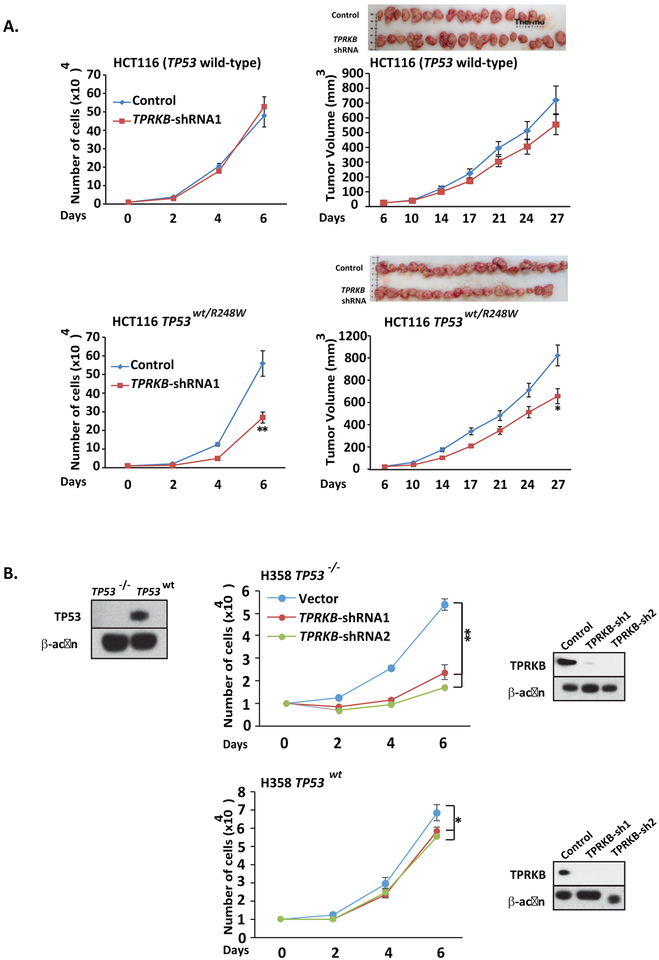
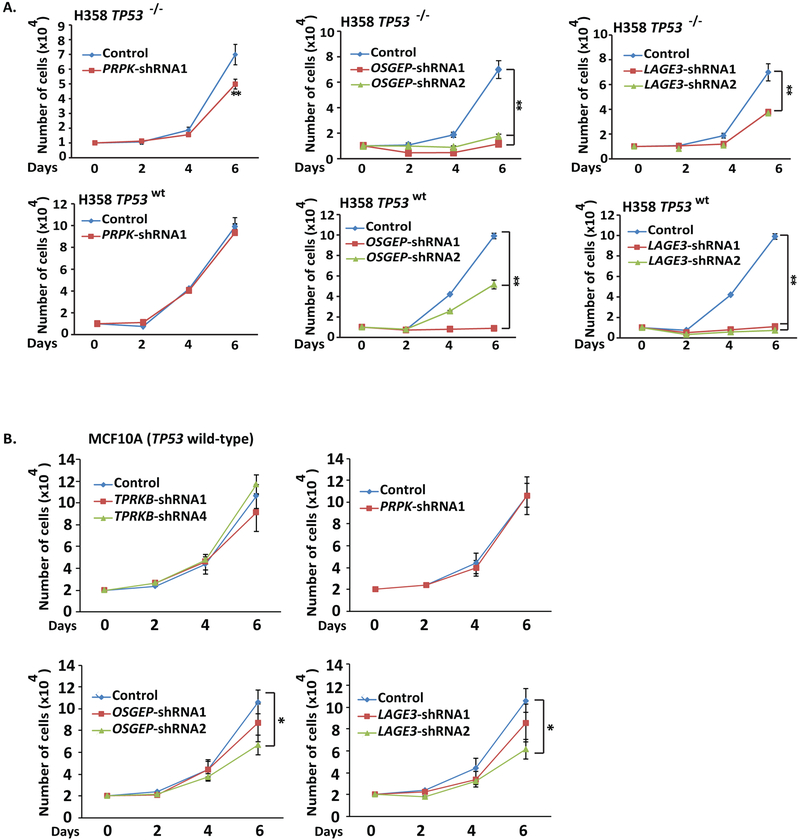
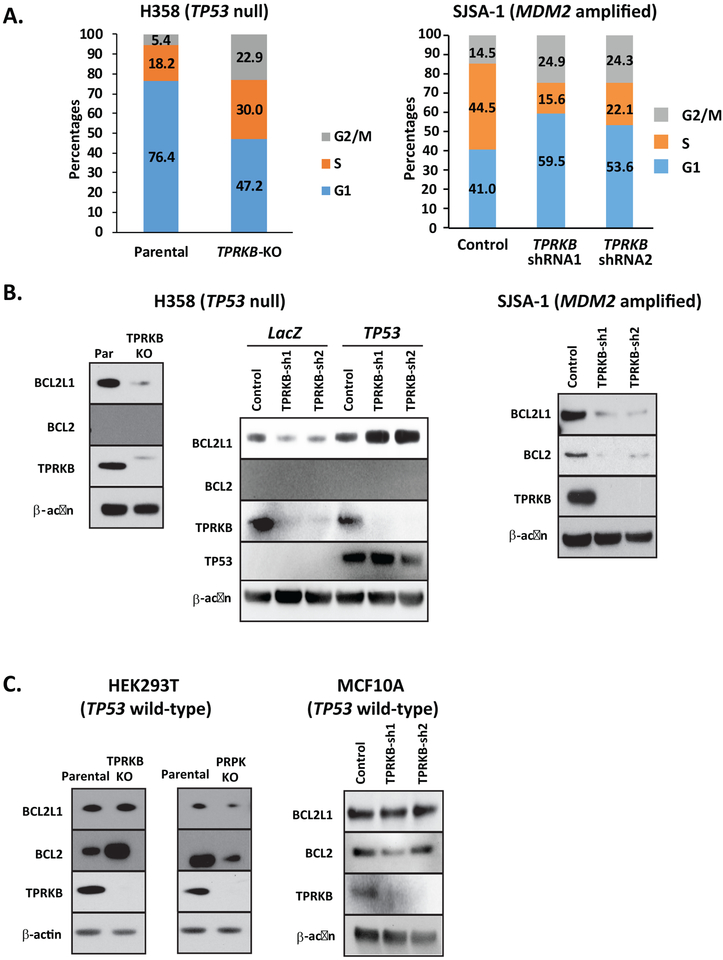
Tumor protein 53 (TP53; p53) is the most frequently altered gene in human cancer. Identification of vulnerabilities imposed by TP53 alterations may enable effective therapeutic approaches. Through analyzing short hairpin RNA (shRNA) screening data, we identified TP53RK-Binding Protein (TPRKB), a poorly characterized member of the tRNA-modifying EKC/KEOPS complex, as the most significant vulnerability in TP53-mutated cancer cell lines. In vitro and in vivo, across multiple benign-immortalized and cancer cell lines, we confirmed that TPRKB knockdown in TP53-deficient cells significantly inhibited proliferation, with minimal effect in TP53 wild-type cells. TP53 reintroduction into TP53-null cells resulted in loss of TPRKB sensitivity, confirming the importance of TP53 status in this context. In addition, cell lines with mutant TP53 or amplified MDM2 (E3-ubiquitin ligase for TP53) also showed high sensitivity to TPRKB knockdown, consistent with TPRKB dependence in a wide array of TP53-altered cancers. Depletion of other EKC/KEOPS complex members exhibited TP53-independent effects, supporting complex-independent functions of TPRKB. Finally, we found that TP53 indirectly mediates TPRKB degradation, which was rescued by coexpression of PRPK, an interacting member of the EKC/KEOPS complex, or proteasome inhibition. Together, these results identify a unique and specific requirement of TPRKB in a variety of TP53-deficient cancers. IMPLICATIONS: Cancer cells with genomic alterations in TP53 are dependent on TPRKB.
©2019 American Association for Cancer Research.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
