

The Bacteriophages of Streptococcus pyogenes
- PMID: 31111820
- PMCID: PMC11314938
- DOI: 10.1128/microbiolspec.GPP3-0059-2018
The Bacteriophages of Streptococcus pyogenes
Abstract
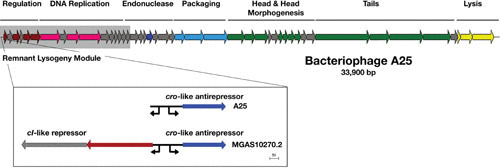
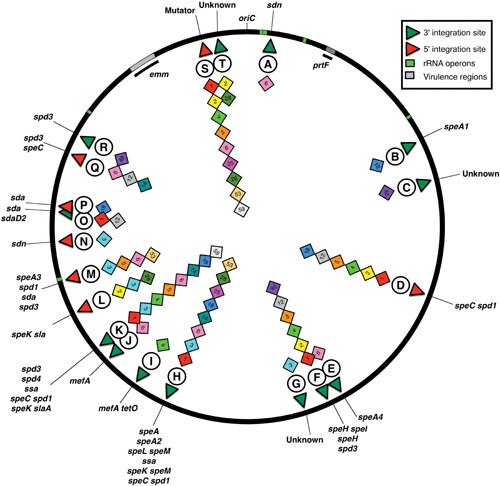
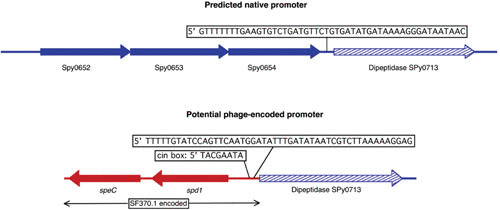
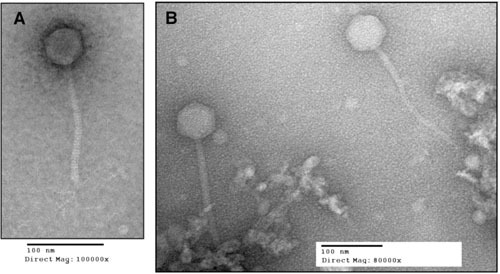
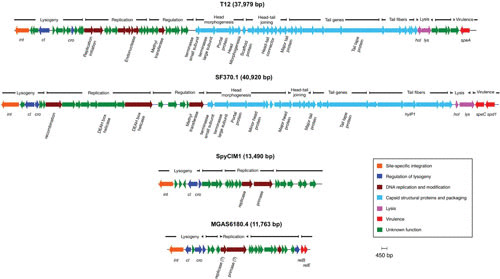
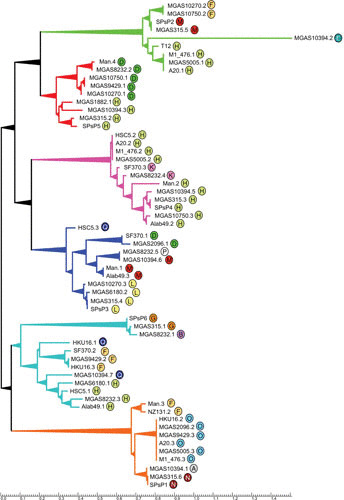
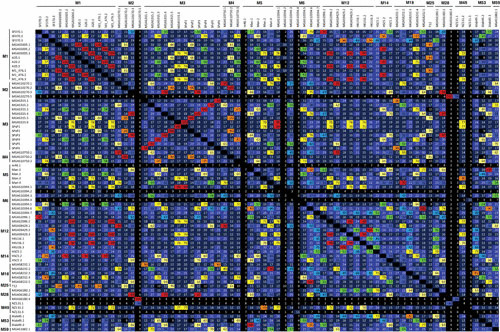
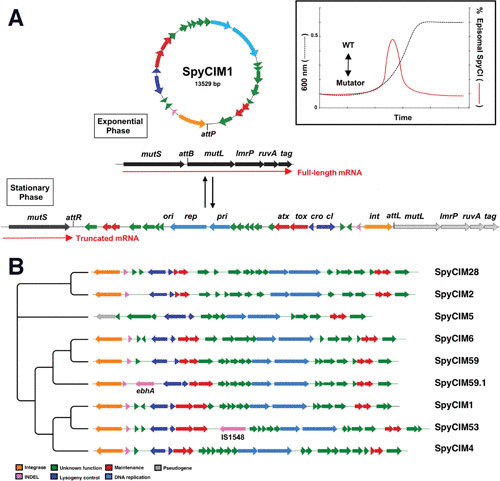
The bacteriophages of Streptococcus pyogenes (group A streptococcus) play a key role in population shaping, genetic transfer, and virulence of this bacterial pathogen. Lytic phages like A25 can alter population distributions through elimination of susceptible serotypes but also serve as key mediators for genetic transfer of virulence genes and antibiotic resistance via generalized transduction. The sequencing of multiple S. pyogenes genomes has uncovered a large and diverse population of endogenous prophages that are vectors for toxins and other virulence factors and occupy multiple attachment sites in the bacterial genomes. Some of these sites for integration appear to have the potential to alter the bacterial phenotype through gene disruption. Remarkably, the phage-like chromosomal islands (SpyCI), which share many characteristics with endogenous prophages, have evolved to mediate a growth-dependent mutator phenotype while acting as global transcriptional regulators. The diverse population of prophages appears to share a large pool of genetic modules that promotes novel combinations that may help disseminate virulence factors to different subpopulations of S. pyogenes. The study of the bacteriophages of this pathogen, both lytic and lysogenic, will continue to be an important endeavor for our understanding of how S. pyogenes continues to be a significant cause of human disease.
Figures

References
-
- Cantacuzene J, Boncieu O. 1926. Modifications subies pare des streptococques d’origine non-scarlatineuse qu contact des produits scarlatineux filtres. C R Acad Sci 182:1185.
-
- Frobisher M, Brown JH. 1927. Transmissible toxicogenicity of streptococci. Bull Johns Hopkins Hosp 41:167–173.
-
- Evans AC. 1933. Inactivation of antistreptococcus bacteriophage by animal fluids. Public Health Rep 48:411–426 10.2307/4580757. - DOI
-
- Evans AC. 1934. Streptococcus bacteriophage: a study of four serological types. Public Health Rep 49:1386–1401 10.2307/4581377. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
