

Tie2 regulates endocardial sprouting and myocardial trabeculation
- PMID: 31112136
- PMCID: PMC6629240
- DOI: 10.1172/jci.insight.96002
Tie2 regulates endocardial sprouting and myocardial trabeculation
Abstract
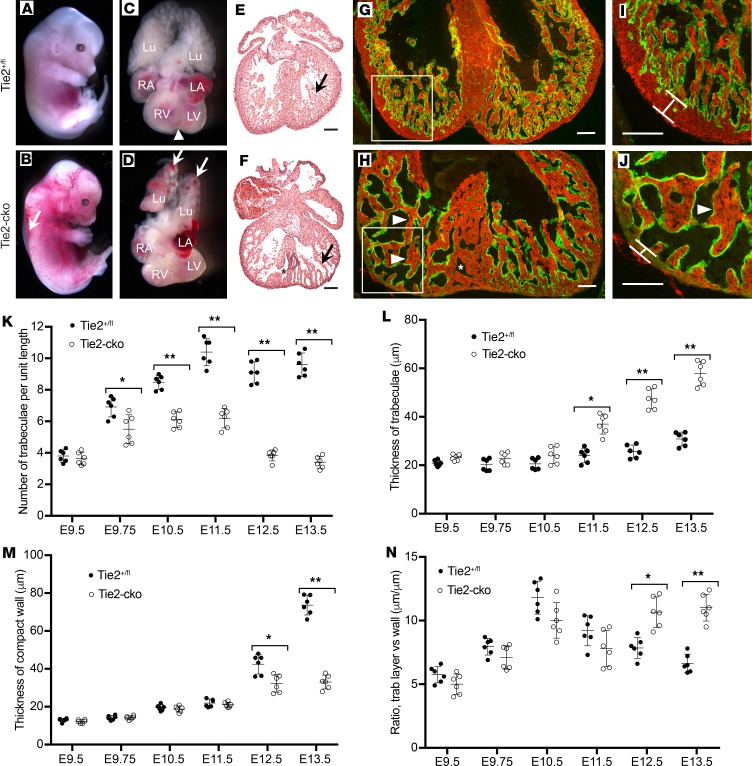
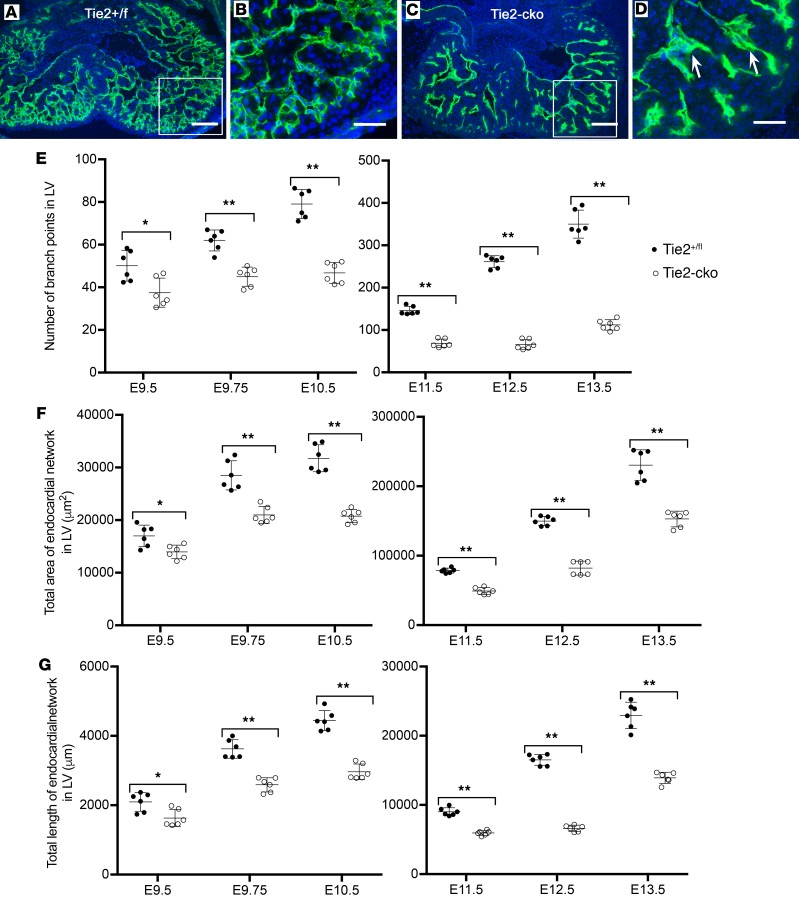
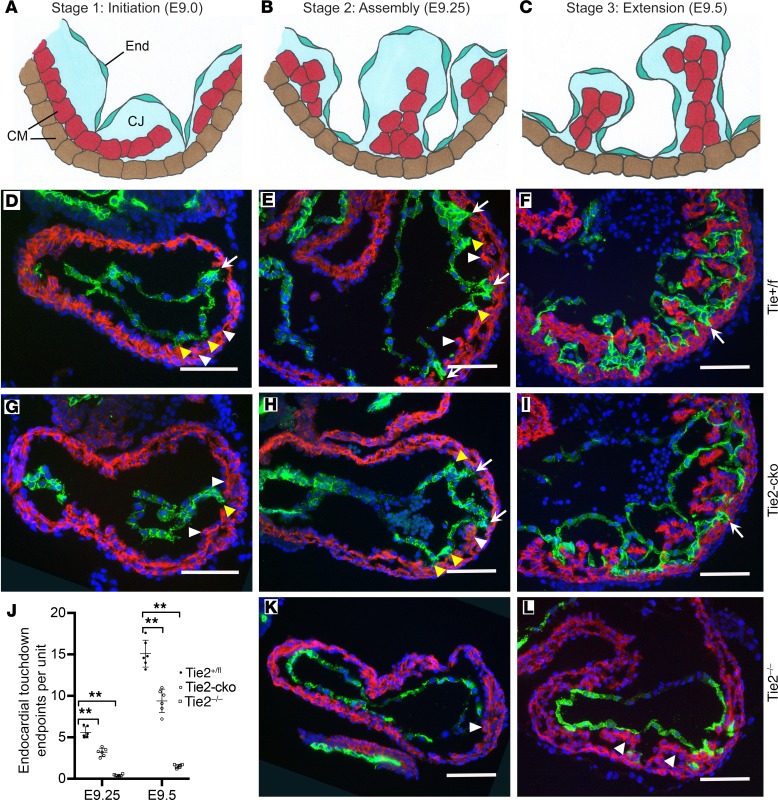
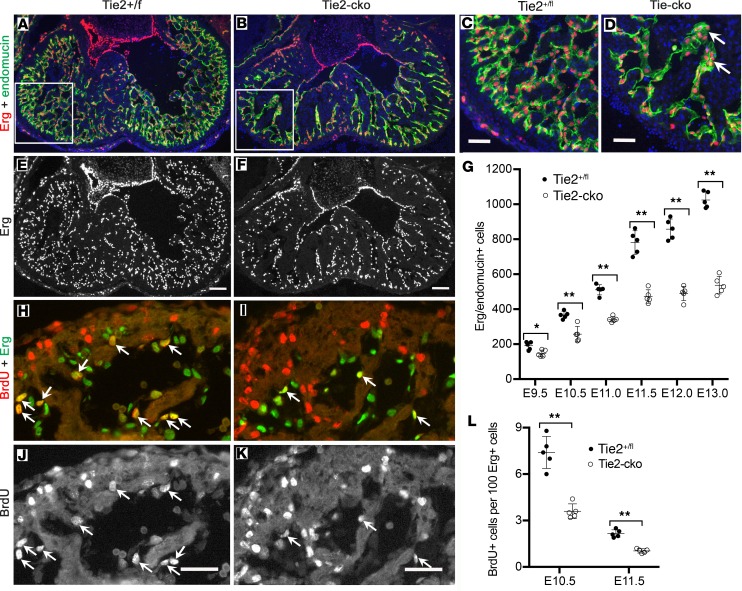
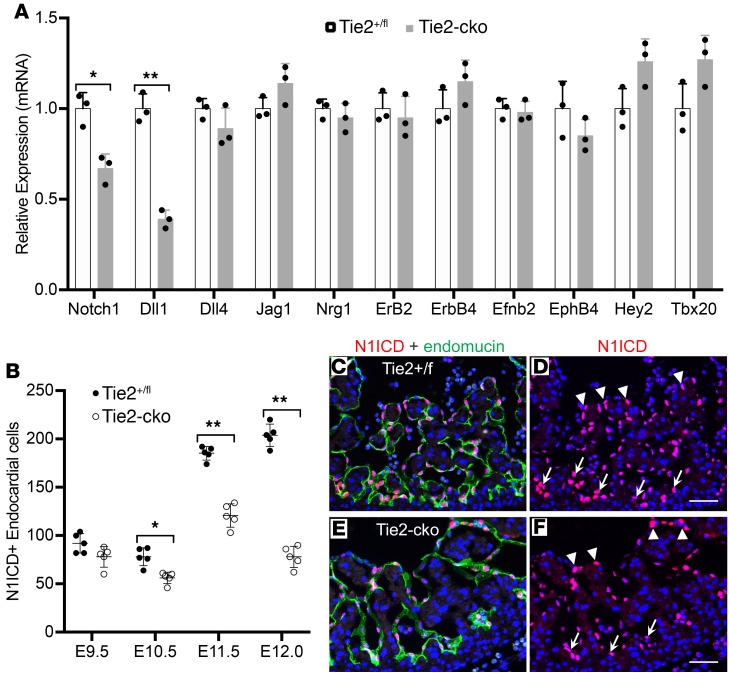
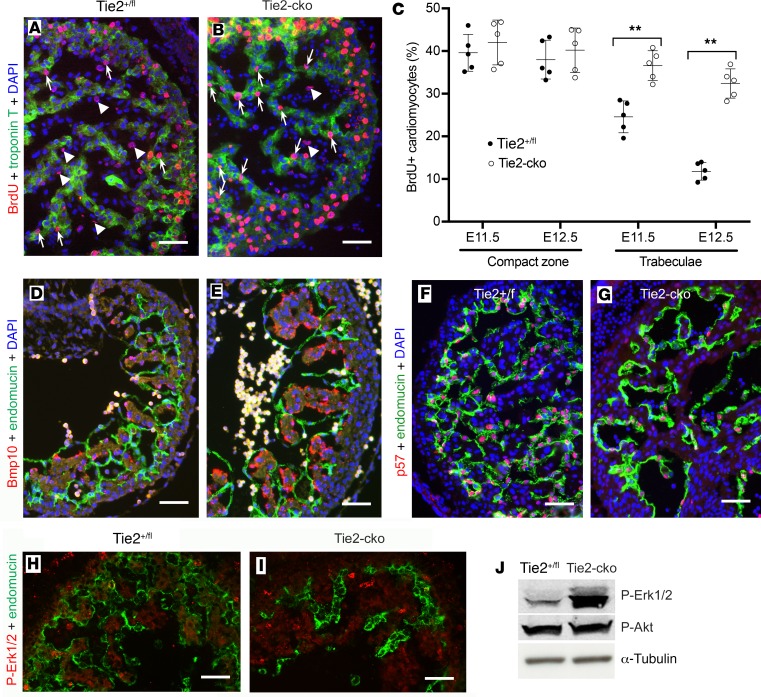
The ang1-Tie2 pathway is required for normal vascular development, but its molecular effectors are not well-defined during cardiac ontogeny. Here we show that endocardial specific attenuation of Tie2 results in mid-gestation lethality due to heart defects associated with a hyperplastic but simplified trabecular meshwork (fewer but thicker trabeculae). Reduced proliferation and production of endocardial cells (ECs) following endocardial loss of Tie2 results in decreased endocardial sprouting required for trabecular assembly and extension. The hyperplastic trabeculae result from enhanced proliferation of trabecular cardiomyocyte (CMs), which is associated with upregulation of Bmp10, increased retinoic acid (RA) signaling, and Erk1/2 hyperphosphorylation in the myocardium. Intriguingly, myocardial phenotypes in Tie2-cko hearts could be partially rescued by inhibiting in utero RA signaling with pan-retinoic acid receptor antagonist BMS493. These findings reveal two complimentary functions of endocardial Tie2 during ventricular chamber formation: ensuring normal trabeculation by supporting EC proliferation and sprouting, and preventing hypertrabeculation via suppression of RA signaling in trabecular CMs.
Keywords: Cardiology; Cardiovascular disease; Development; Embryonic development; Mouse models.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
