

Herpes Simplex Virus Evasion of Early Host Antiviral Responses
- PMID: 31114761
- PMCID: PMC6503643
- DOI: 10.3389/fcimb.2019.00127
Herpes Simplex Virus Evasion of Early Host Antiviral Responses
Abstract
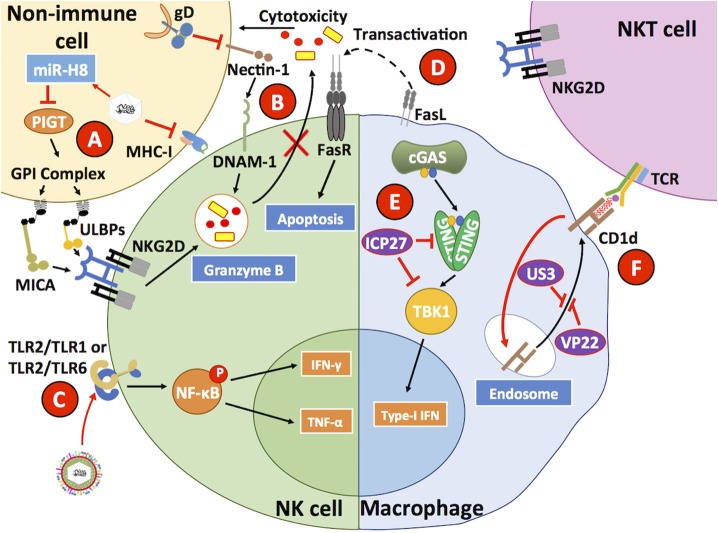
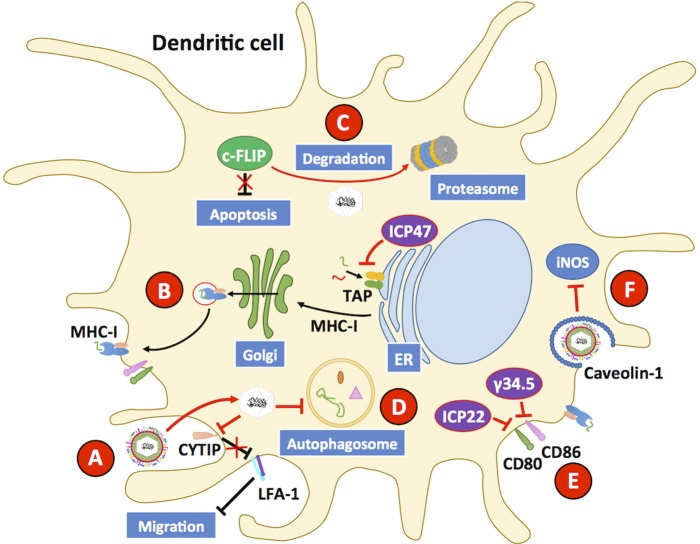
Herpes simplex viruses type 1 (HSV-1) and type 2 (HSV-2) have co-evolved with humans for thousands of years and are present at a high prevalence in the population worldwide. HSV infections are responsible for several illnesses including skin and mucosal lesions, blindness and even life-threatening encephalitis in both, immunocompetent and immunocompromised individuals of all ages. Therefore, diseases caused by HSVs represent significant public health burdens. Similar to other herpesviruses, HSV-1 and HSV-2 produce lifelong infections in the host by establishing latency in neurons and sporadically reactivating from these cells, eliciting recurrences that are accompanied by viral shedding in both, symptomatic and asymptomatic individuals. The ability of HSVs to persist and recur in otherwise healthy individuals is likely given by the numerous virulence factors that these viruses have evolved to evade host antiviral responses. Here, we review and discuss molecular mechanisms used by HSVs to evade early innate antiviral responses, which are the first lines of defense against these viruses. A comprehensive understanding of how HSVs evade host early antiviral responses could contribute to the development of novel therapies and vaccines to counteract these viruses.
Keywords: apoptosis; cytosolic nucleic acid receptors; dendritic cells (DCs); inflammasome; innate immunity; interferon (IFN); natural killer cells (NK cells); toll-like receptors (TLRs).
Figures
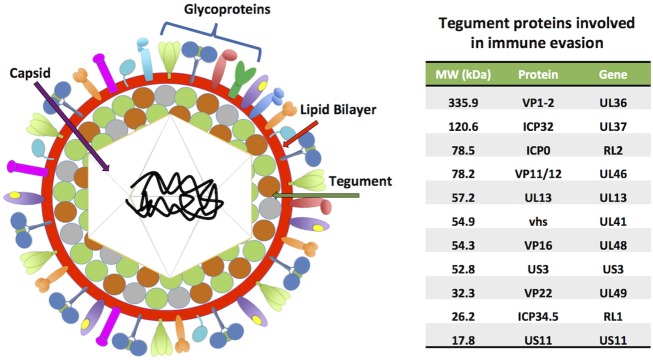
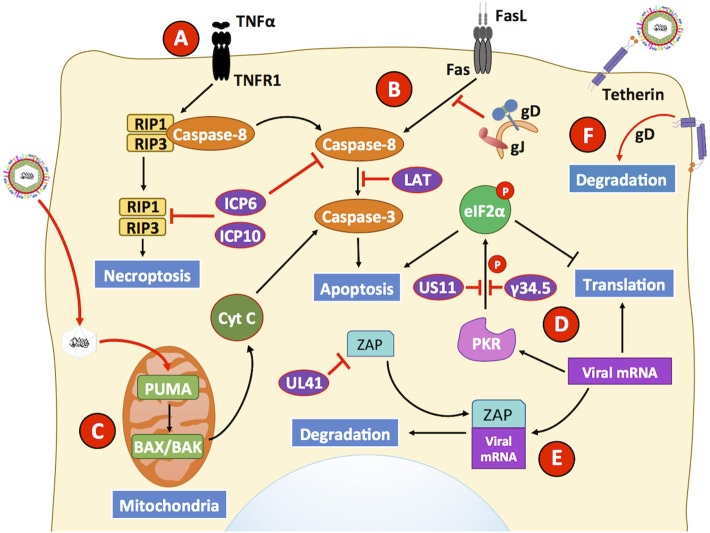
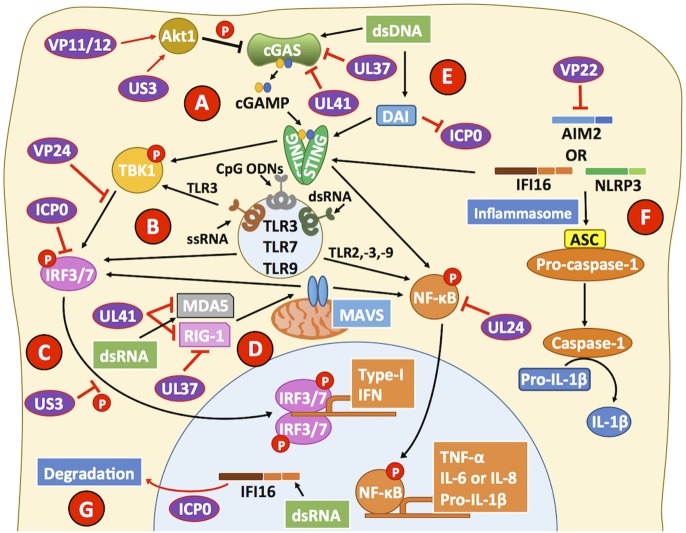
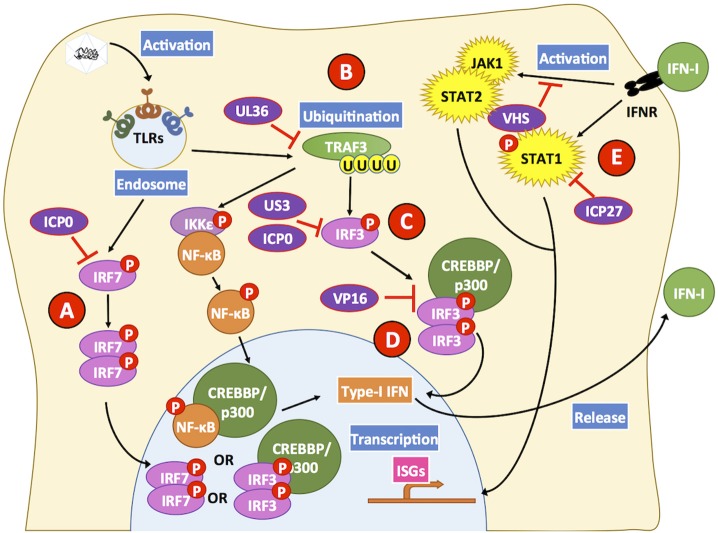
References
-
- Al-Khatib K., Williams B. R., Silverman R. H., Halford W., Carr D. J. (2004). Distinctive roles for 2',5'-oligoadenylate synthetases and double-stranded RNA-dependent protein kinase R in the in vivo antiviral effect of an adenoviral vector expressing murine IFN-beta. J. Immunol. 172, 5638–5647. 10.4049/jimmunol.172.9.5638 - DOI - PMC - PubMed
-
- Ansari M. A., Dutta S., Veettil M. V., Dutta D., Iqbal J., Kumar B., et al. (2015). Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-beta responses. PLoS Pathog. 11:e1005019. 10.1371/journal.ppat.1005019 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources