

ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer
- PMID: 31117237
- PMCID: PMC6566596
- DOI: 10.3390/ijms20102505
ERK is a Pivotal Player of Chemo-Immune-Resistance in Cancer
Abstract
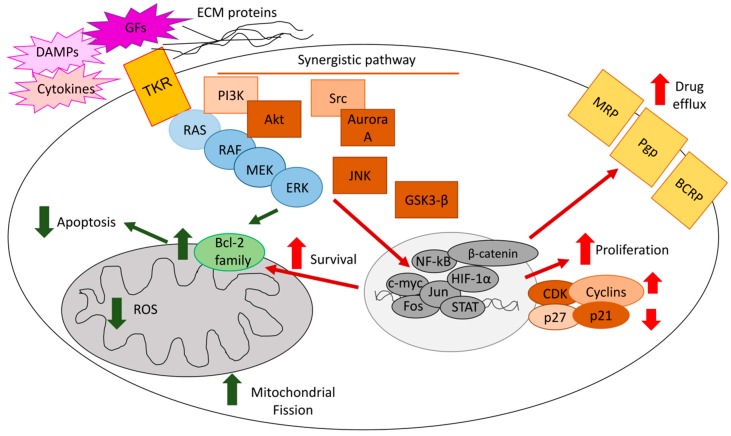
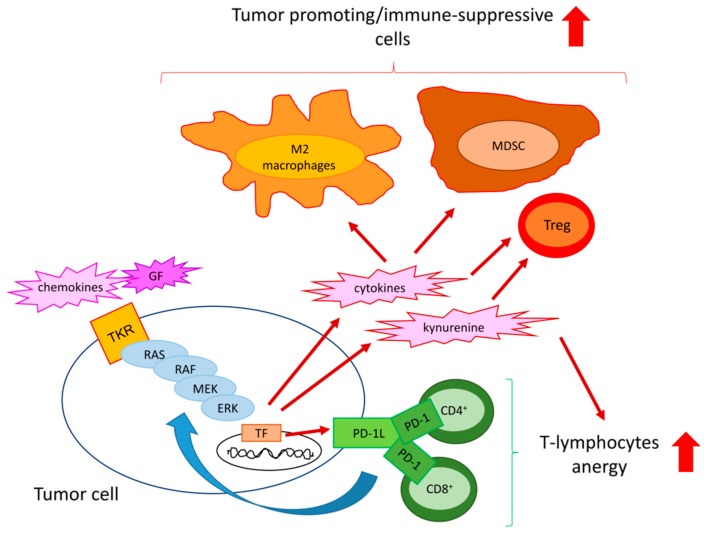
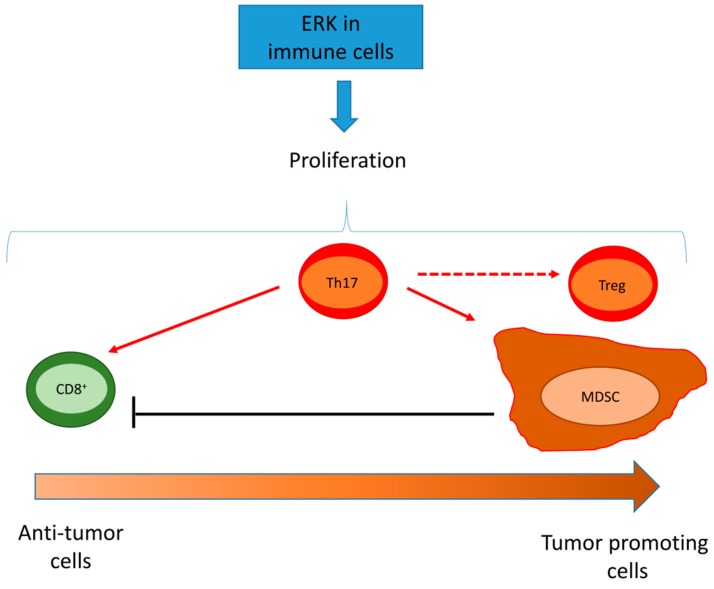
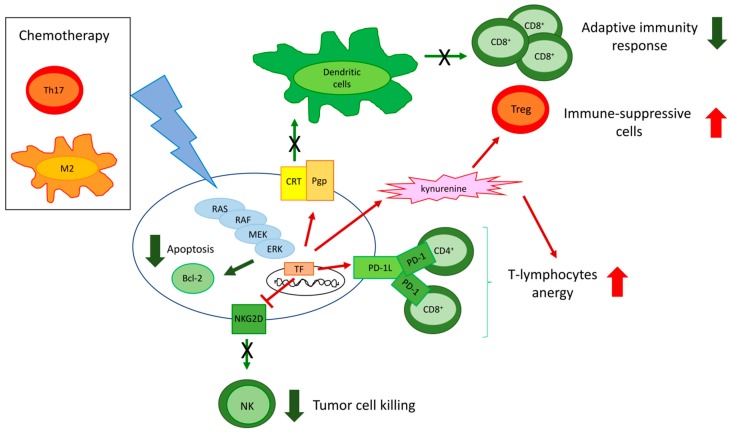
The extracellular signal-related kinases (ERKs) act as pleiotropic molecules in tumors, where they activate pro-survival pathways leading to cell proliferation and migration, as well as modulate apoptosis, differentiation, and senescence. Given its central role as sensor of extracellular signals, ERK transduction system is widely exploited by cancer cells subjected to environmental stresses, such as chemotherapy and anti-tumor activity of the host immune system. Aggressive tumors have a tremendous ability to adapt and survive in stressing and unfavorable conditions. The simultaneous resistance to chemotherapy and immune system responses is common, and ERK signaling plays a key role in both types of resistance. In this review, we dissect the main ERK-dependent mechanisms and feedback circuitries that simultaneously determine chemoresistance and immune-resistance/immune-escape in cancer cells. We discuss the pros and cons of targeting ERK signaling to induce chemo-immune-sensitization in refractory tumors.
Keywords: ERK; chemoresistance; immune-escape; immune-resistance.
Conflict of interest statement
The authors declare no conflict of interest. The founding sponsors had no role in the writing of the manuscript and in the decision to publish it.
Figures
References
-
- Megiorni F., Camero S., Ceccarelli S., McDowell H.P., Mannarino O., Marampon F., Pizer B., Shukla R., Pizzuti A., Marchese C., et al. DNMT3B in vitro knocking-down is able to reverse embryonal rhabdomyosarcoma cell phenotype through inhibition of proliferation and induction of myogenic differentiation. Oncotarget. 2016;7:79342–79356. doi: 10.18632/oncotarget.12688. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
