

Rapid Structure Determination of Molecular Solids Using Chemical Shifts Directed by Unambiguous Prior Constraints
- PMID: 31117663
- PMCID: PMC7540916
- DOI: 10.1021/jacs.9b03908
Rapid Structure Determination of Molecular Solids Using Chemical Shifts Directed by Unambiguous Prior Constraints
Abstract
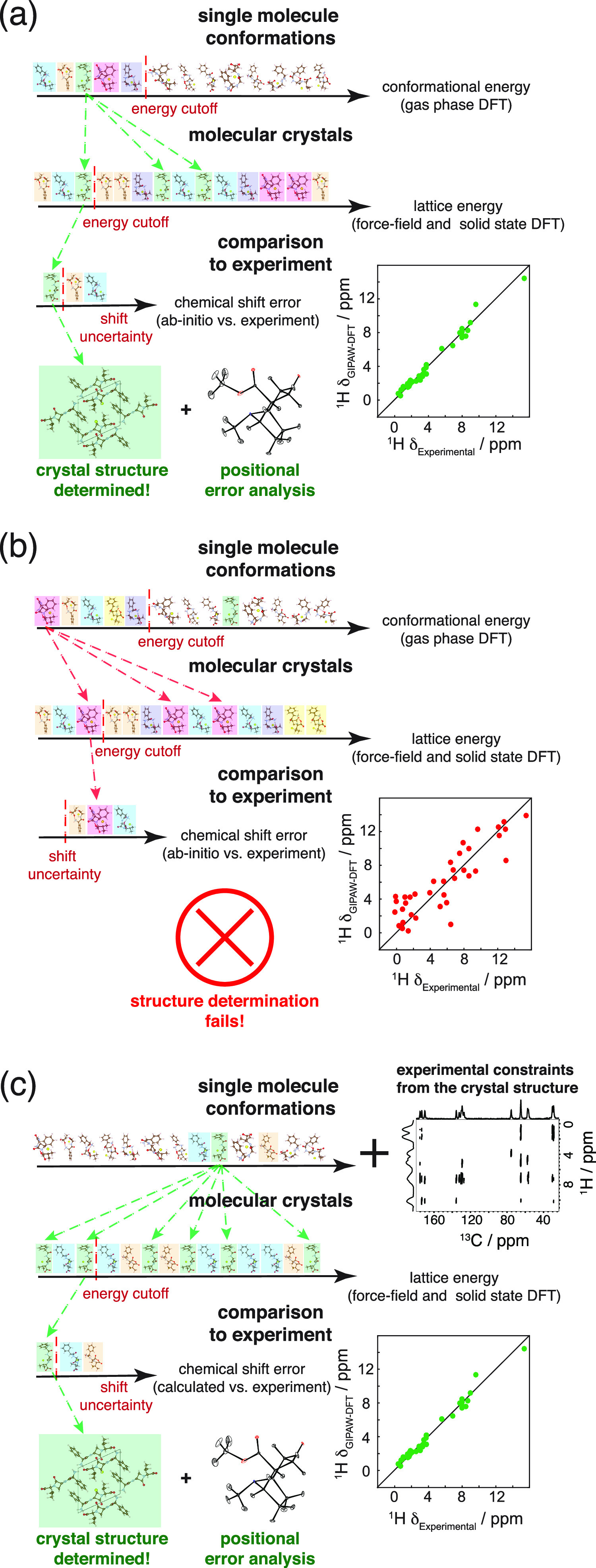
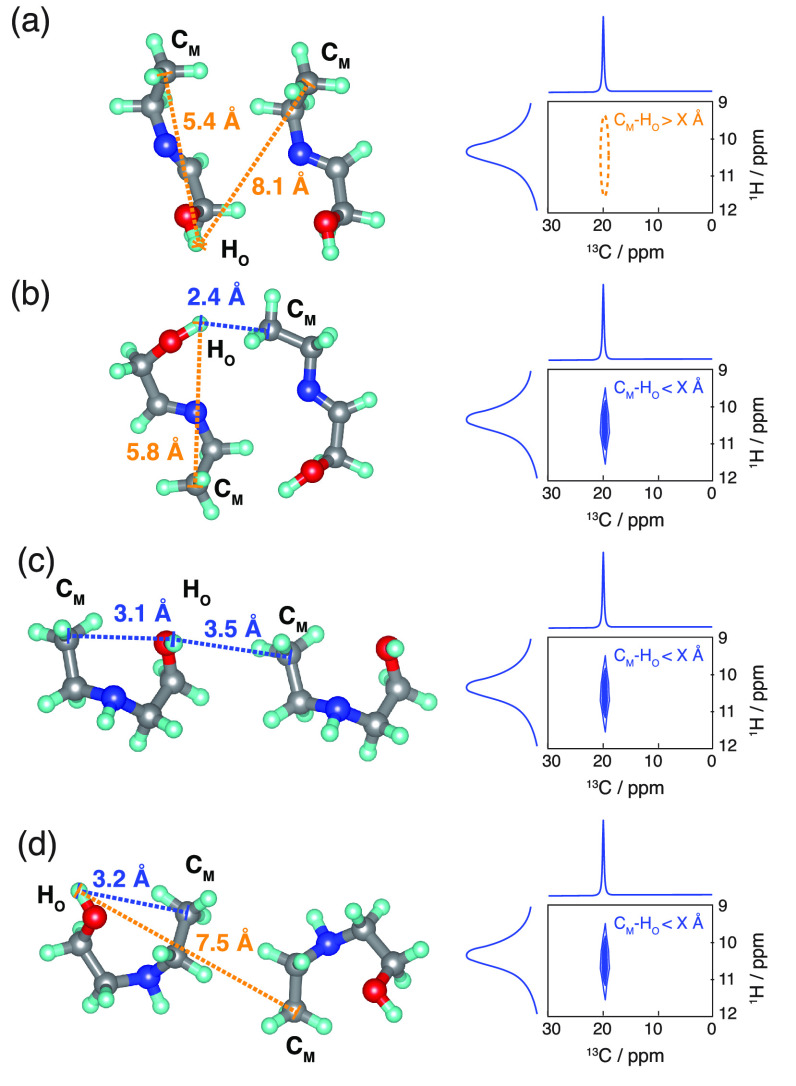
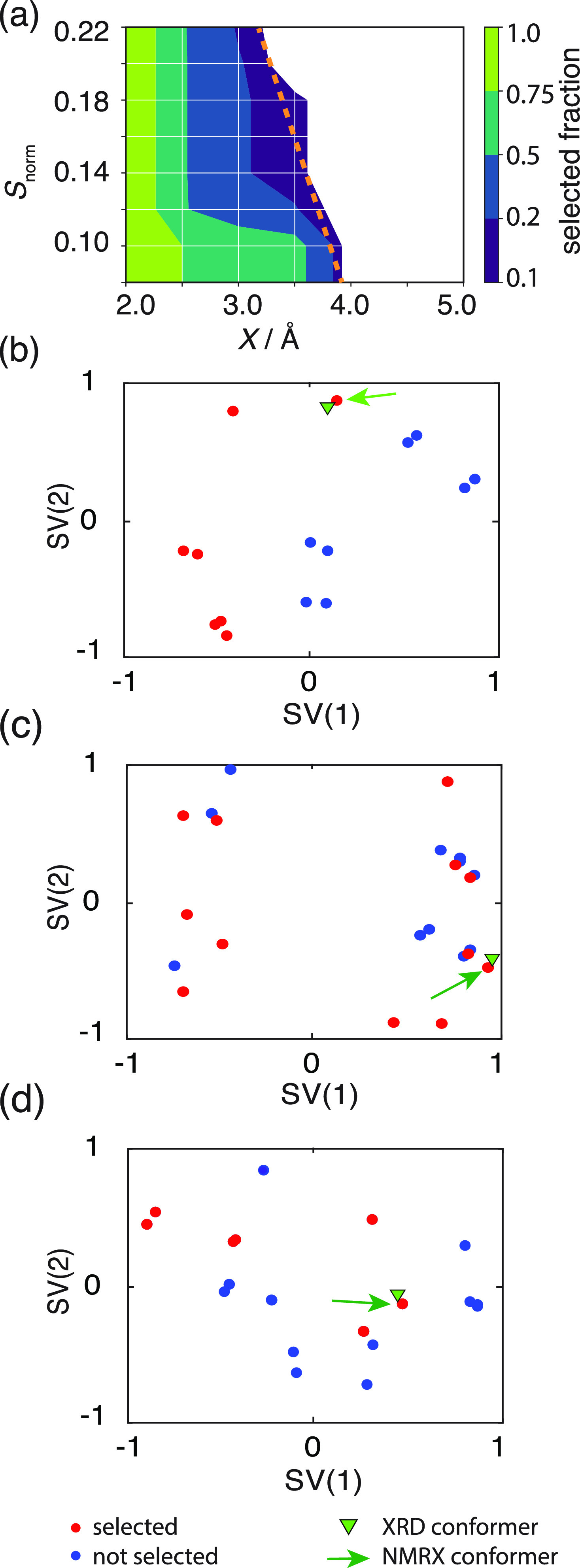
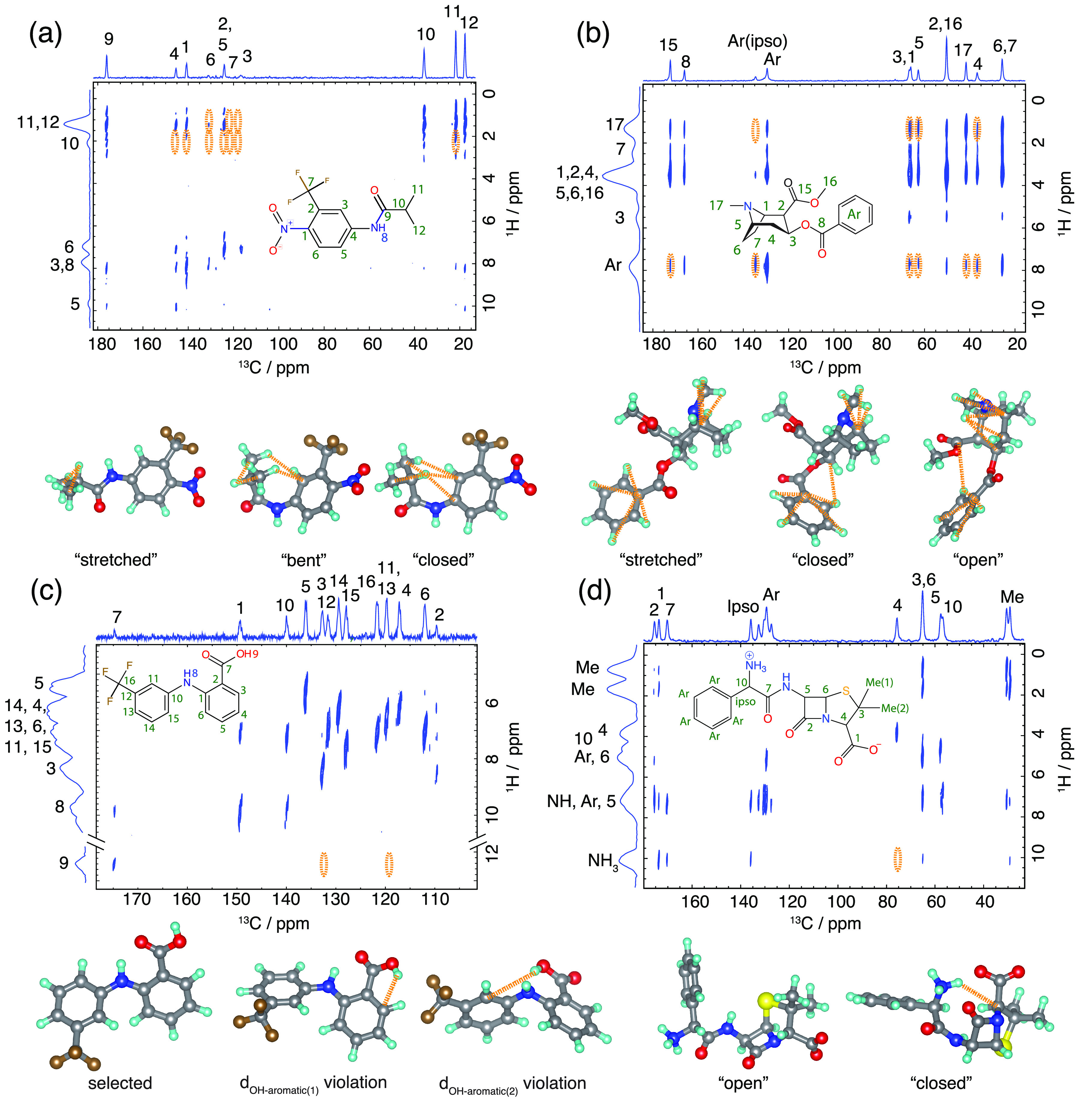
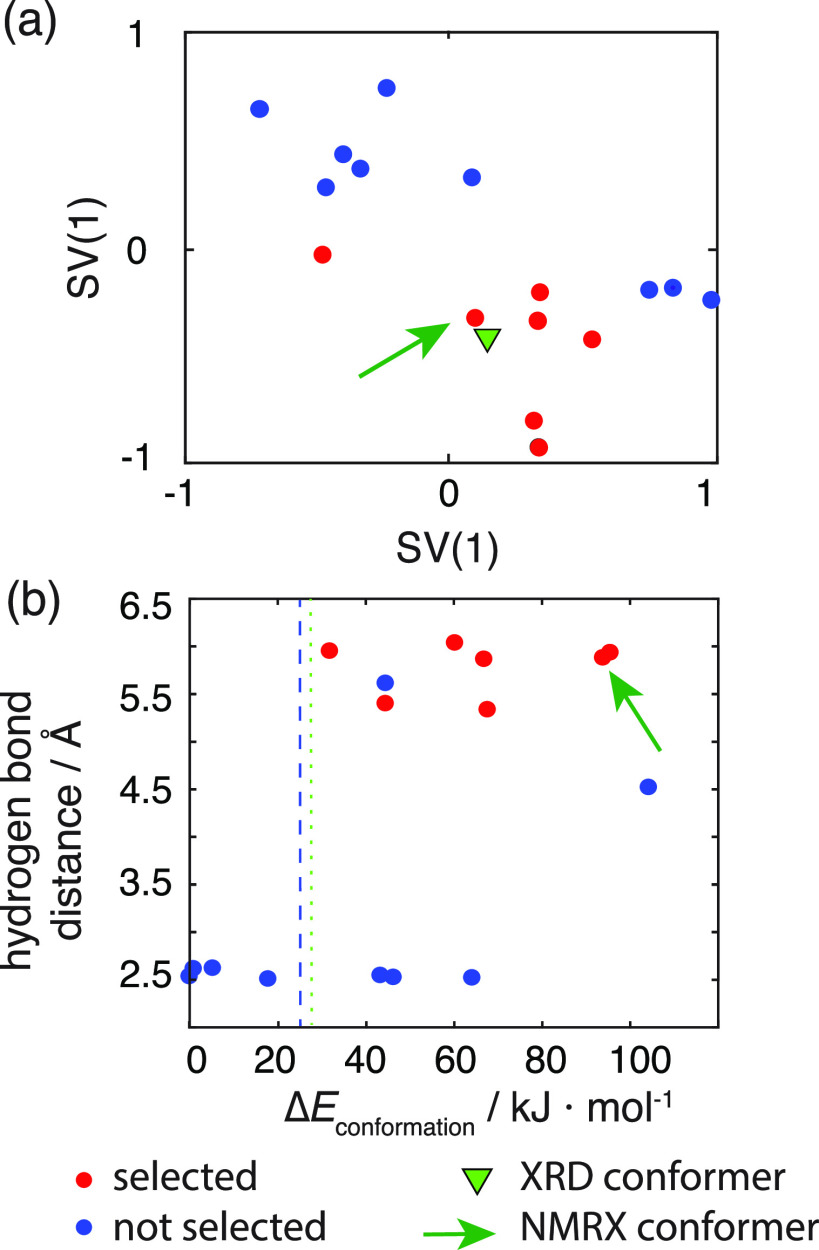
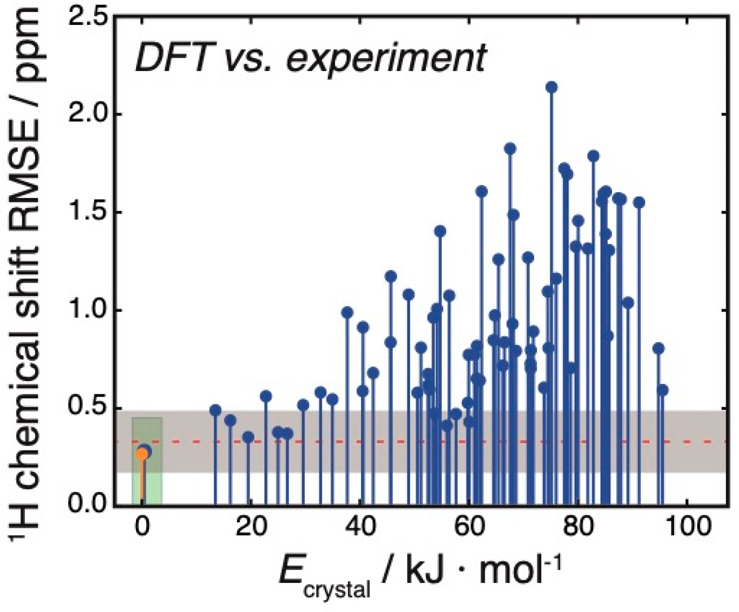
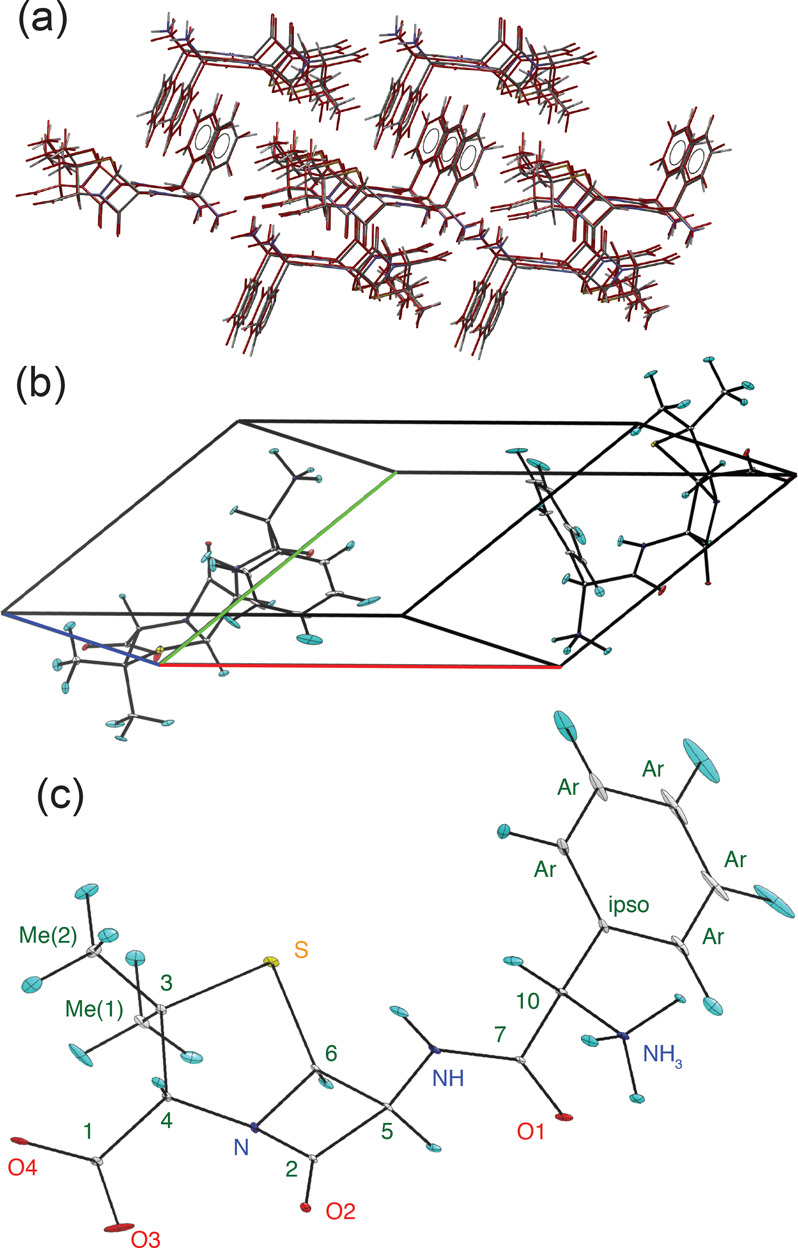
NMR-based crystallography approaches involving the combination of crystal structure prediction methods, ab initio calculated chemical shifts and solid-state NMR experiments are powerful methods for crystal structure determination of microcrystalline powders. However, currently structural information obtained from solid-state NMR is usually included only after a set of candidate crystal structures has already been independently generated, starting from a set of single-molecule conformations. Here, we show with the case of ampicillin that this can lead to failure of structure determination. We propose a crystal structure determination method that includes experimental constraints during conformer selection. In order to overcome the problem that experimental measurements on the crystalline samples are not obviously translatable to restrict the single-molecule conformational space, we propose constraints based on the analysis of absent cross-peaks in solid-state NMR correlation experiments. We show that these absences provide unambiguous structural constraints on both the crystal structure and the gas-phase conformations, and therefore can be used for unambiguous selection. The approach is parametrized on the crystal structure determination of flutamide, flufenamic acid, and cocaine, where we reduce the computational cost by around 50%. Most importantly, the method is then shown to correctly determine the crystal structure of ampicillin, which would have failed using current methods because it adopts a high-energy conformer in its crystal structure. The average positional RMSE on the NMR powder structure is ⟨rav⟩ = 0.176 Å, which corresponds to an average equivalent displacement parameter Ueq = 0.0103 Å2.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Grazulis S.; Chateigner D.; Downs R. T.; Yokochi A. F.; Quiros M.; Lutterotti L.; Manakova E.; Butkus J.; Moeck P.; Le Bail A. Crystallography Open Database - an open-access collection of crystal structures. J. Appl. Crystallogr. 2009, 42 (4), 726–729. 10.1107/S0021889809016690. - DOI - PMC - PubMed
-
- Grazulis S.; Daskevic A.; Merkys A.; Chateigner D.; Lutterotti L.; Quiros M.; Serebryanaya N. R.; Moeck P.; Downs R. T.; Le Bail A. Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res. 2012, 40 (D1), D420–D427. 10.1093/nar/gkr900. - DOI - PMC - PubMed
-
- Brown I. D.; Bergerhoff G. Databases of Inorganic Crystal-Structures. Abstr. Pap. Am. Chem. Soc. 1979, (Sep), 36–36.
-
- Batisai E.; Ayamine A.; Kilinkissa O. E. Y.; Báthori N. B. Melting point–solubility–structure correlations in multicomponent crystals containing fumaric or adipic acid. CrystEngComm 2014, 16 (43), 9992–9998. 10.1039/C4CE01298D. - DOI
Publication types
LinkOut - more resources
Full Text Sources

