

An update on genetic frontotemporal dementia
- PMID: 31119452
- PMCID: PMC6647117
- DOI: 10.1007/s00415-019-09363-4
An update on genetic frontotemporal dementia
Abstract
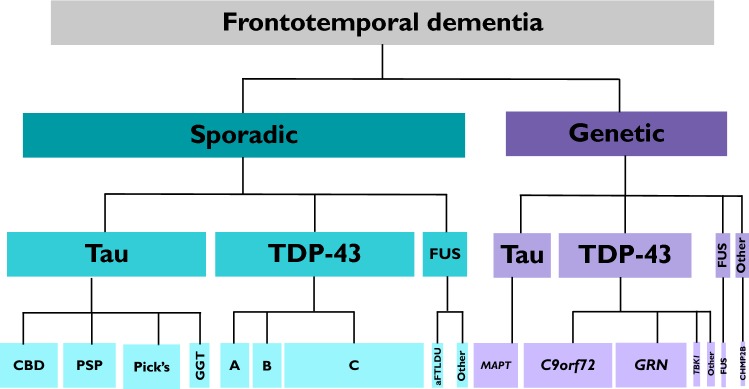
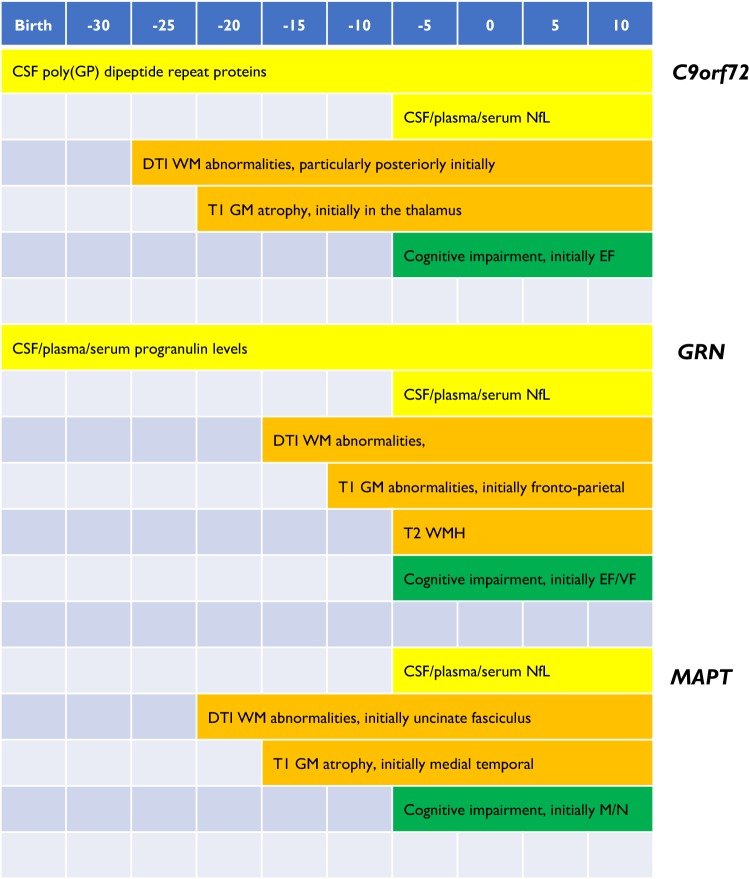
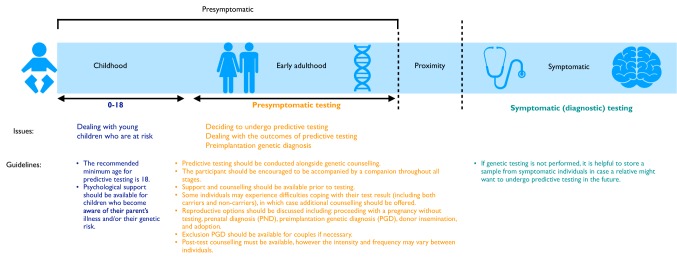
Frontotemporal dementia (FTD) is a highly heritable group of neurodegenerative disorders, with around 30% of patients having a strong family history. The majority of that heritability is accounted for by autosomal dominant mutations in the chromosome 9 open reading frame 72 (C9orf72), progranulin (GRN), and microtubule-associated protein tau (MAPT) genes, with mutations more rarely seen in a number of other genes. This review will discuss the recent updates in the field of genetic FTD. Age at symptom onset in genetic FTD is variable with recently identified genetic modifiers including TMEM106B (in GRN carriers particularly) and a polymorphism at a locus containing two overlapping genes LOC101929163 and C6orf10 (in C9orf72 carriers). Behavioural variant FTD (bvFTD) is the most common diagnosis in each of the genetic groups, although in C9orf72 carriers amyotrophic lateral sclerosis either alone, or with bvFTD, is also common. An atypical neuropsychiatric presentation is also seen in C9orf72 carriers and family members of carriers are at greater risk of psychiatric disorders including schizophrenia and autistic spectrum disorders. Large natural history studies of presymptomatic genetic FTD are now underway both in Europe/Canada (GENFI-the Genetic FTD Initiative) and in the US (ARTFL/LEFFTDS study), collaborating together under the banner of the FTD Prevention Initiative (FPI). These studies are taking forward the validation of cognitive, imaging and fluid biomarkers that aim to robustly measure disease onset, staging and progression in genetic FTD. Grey matter changes on MRI and hypometabolism on FDG-PET are seen at least 10 years before symptom onset with white matter abnormalities seen earlier, but the pattern and exact timing of changes differ between different genetic groups. In contrast, tau PET has yet to show promise in genetic FTD. Three key fluid biomarkers have been identified so far that are likely to be helpful in clinical trials-CSF or blood neurofilament light chain levels (in all groups), CSF or blood progranulin levels (in GRN carriers) and CSF poly(GP) dipeptide repeat protein levels (in C9orf72 carriers). Increased knowledge about genetic FTD has led to more clinical presymptomatic genetic testing but this has not yet been mirrored in the development of either an accepted FTD-specific testing protocol or provision of appropriate psychological support mechanisms for those living through the at-risk phase. This will become even more relevant as disease-modifying therapy trials start in each of the genetic groups over the next few years.
Keywords: Biomarkers; C9orf72; Frontotemporal dementia; Neurogenetics; Progranulin; Tau.
Conflict of interest statement
We have no conflicts of interest.
Figures
References
-
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EG, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini ML, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(Pt 9):2456–2477. - PMC - PubMed
-
- Gorno-Tempini ML, Hillis AE, Weintraub S, Kertesz A, Mendez M, Cappa SF, Ogar JM, Rohrer JD, Black S, Boeve BF, Manes F, Dronkers NF, Vandenberghe R, Rascovsky K, Patterson K, Miller BL, Knopman DS, Hodges JR, Mesulam MM, Grossman M. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006–1014. - PMC - PubMed
-
- Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, Mioshi E, Roberts-South A, Benatar M, HortobáGyi T, Rosenfeld J, Silani V, Ince PG, Turner MR. Amyotrophic lateral sclerosis—frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Later Scler Frontotempor Degener. 2017;18(3–4):153–174. - PMC - PubMed
-
- Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Tröster AI, Vidailhet M, Weiner WJ. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496–503. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
