

MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease
- PMID: 31121115
- PMCID: PMC6538307
- DOI: 10.1016/j.cels.2019.04.003
MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease
Abstract
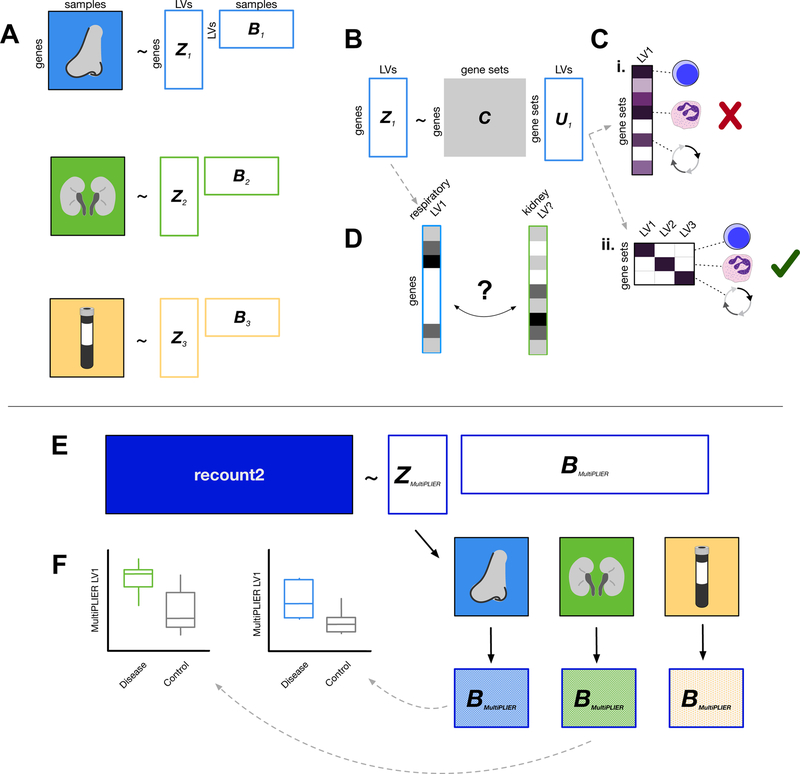
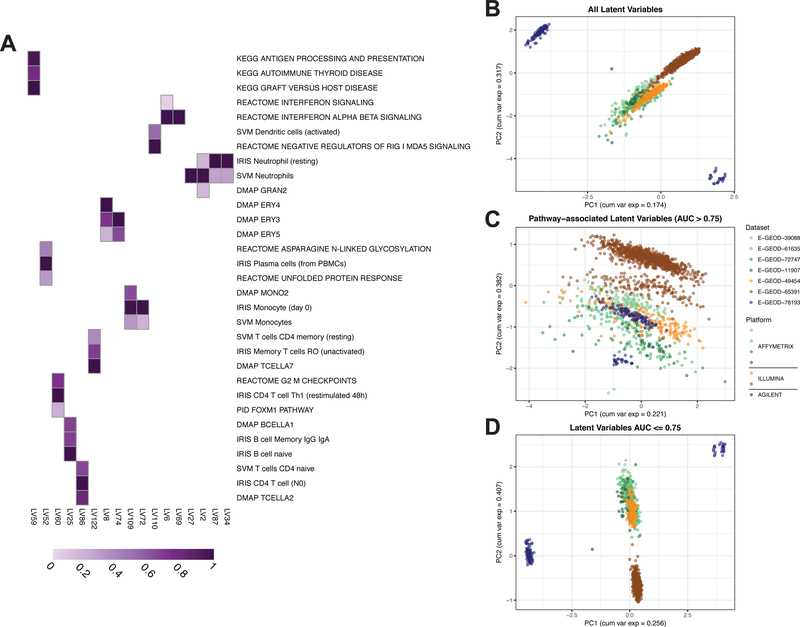
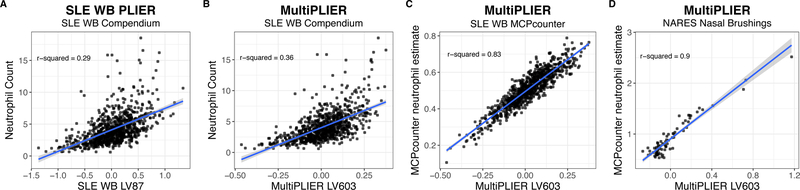
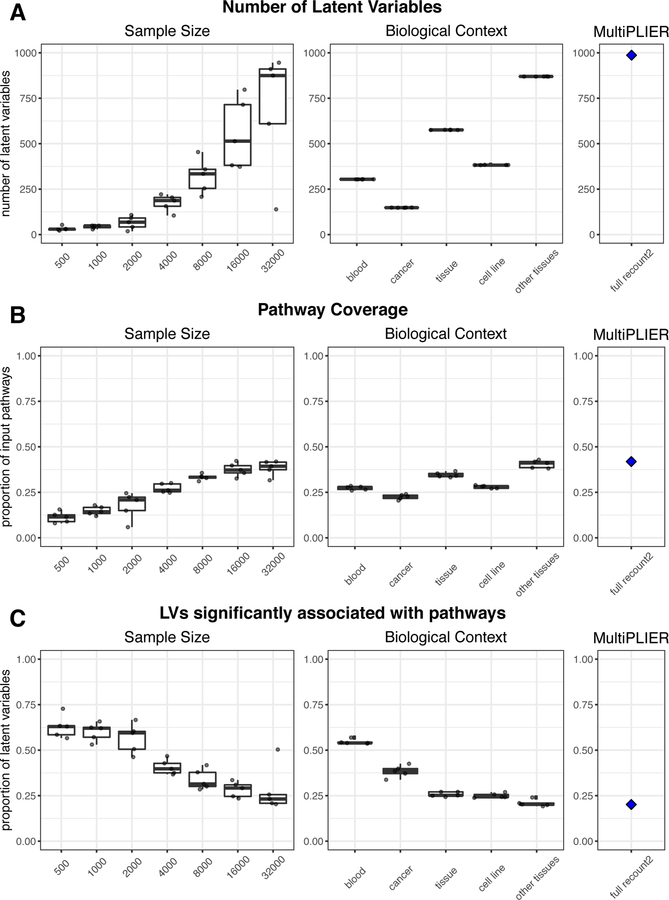
Most gene expression datasets generated by individual researchers are too small to fully benefit from unsupervised machine-learning methods. In the case of rare diseases, there may be too few cases available, even when multiple studies are combined. To address this challenge, we utilize transfer learning to extract coordinated expression patterns and use learned patterns to analyze small rare disease datasets. We trained a pathway-level information extractor (PLIER) model on a large public data compendium comprising multiple experiments, tissues, and biological conditions and then transferred the model to small datasets in an approach we call MultiPLIER. Models constructed from the public data compendium included features that aligned well to known biological factors and were more comprehensive than those constructed from individual datasets or conditions. When transferred to rare disease datasets, the models describe biological processes related to disease severity more effectively than models trained only on a given dataset.
Keywords: genomics; machine learning; medulloblastoma; rare diseases; transcriptomics; transfer learning; unsupervised learning; vasculitis.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
Figures
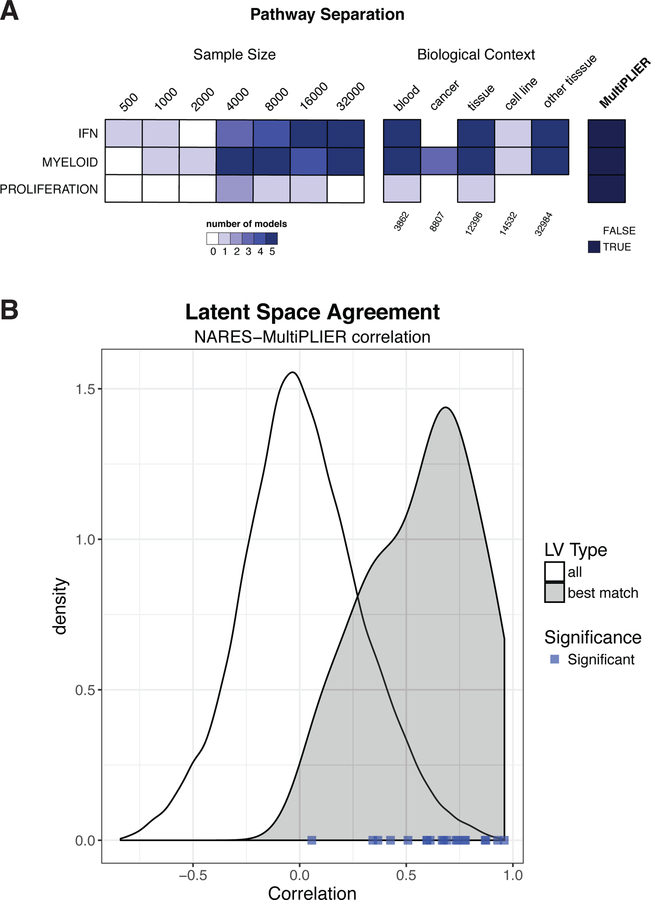
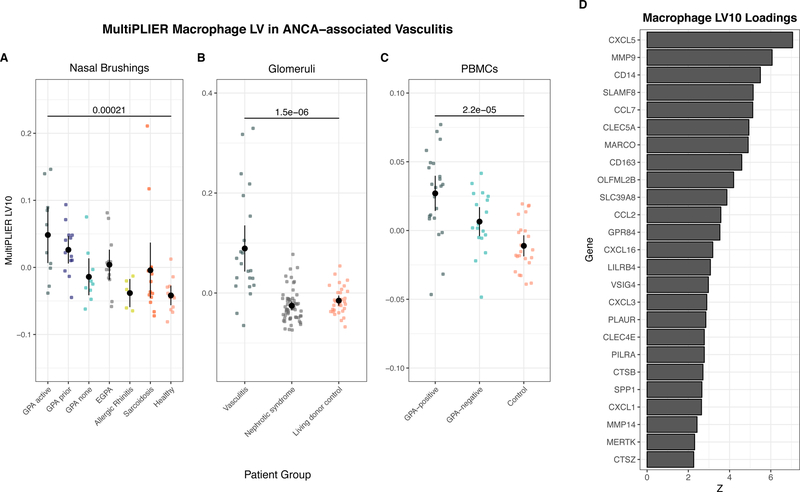
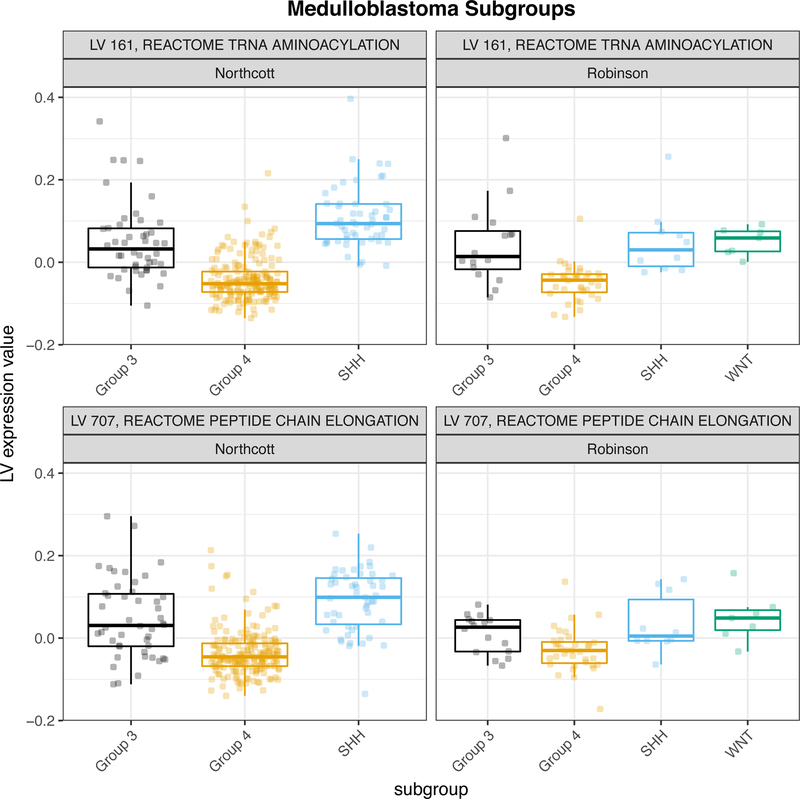
Comment in
-
Evaluation of Taroni et al.: Understanding Rare Diseases by MultiPLIER.Cell Syst. 2019 May 22;8(5):359-360. doi: 10.1016/j.cels.2019.05.001. Cell Syst. 2019. PMID: 31121113
References
-
- Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, Fong S, van Lookeren Campagne M, Godowski P, and Williams PM (2005). Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes and Immunity 6, 319–331. - PubMed
-
- Allen GI, Grosenick L, and Taylor J (2013). A Generalized Least-Square Matrix Decomposition. Journal of the American Statistical Association 109, 145–159.
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
