

Multiplexed primer extension sequencing: A targeted RNA-seq method that enables high-precision quantitation of mRNA splicing isoforms and rare pre-mRNA splicing intermediates
- PMID: 31121301
- PMCID: PMC6868320
- DOI: 10.1016/j.ymeth.2019.05.013
Multiplexed primer extension sequencing: A targeted RNA-seq method that enables high-precision quantitation of mRNA splicing isoforms and rare pre-mRNA splicing intermediates
Abstract
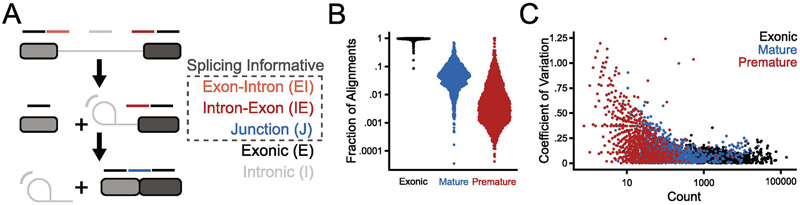
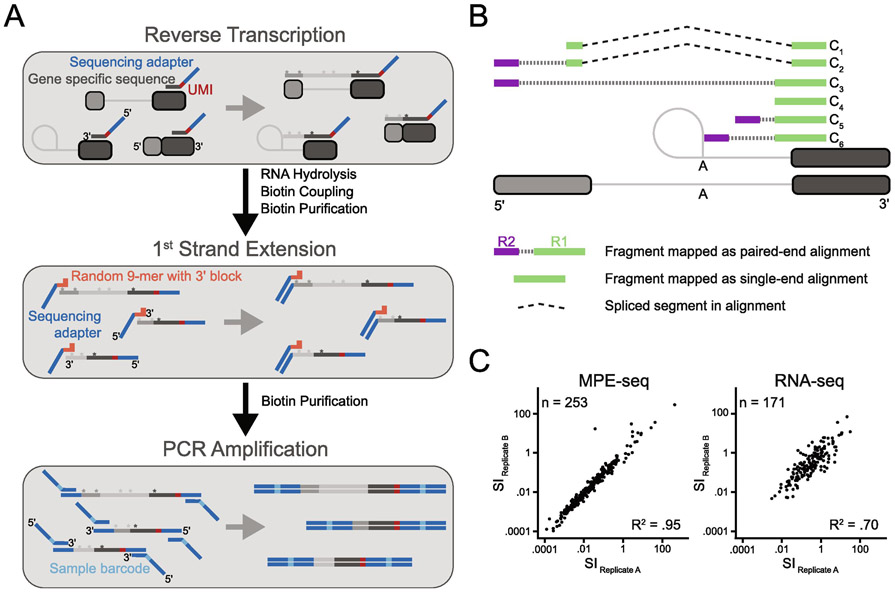
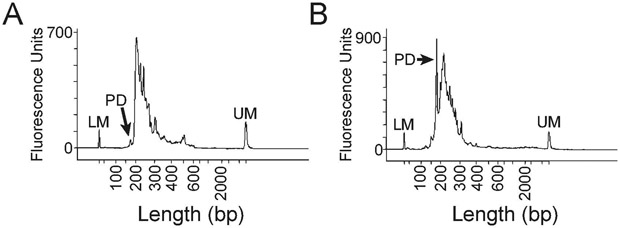
The study of pre-mRNA splicing has been greatly aided by the advent of RNA sequencing (RNA-seq), which enables the genome-wide detection of discrete splice isoforms. Quantification of these splice isoforms requires analysis of splicing informative sequencing reads, those that unambiguously map to a single splice isoform, including exon-intron spanning alignments corresponding to retained introns, as well as exon-exon junction spanning alignments corresponding to either canonically- or alternatively-spliced isoforms. Because most RNA-seq experiments are designed to produce sequencing alignments that uniformly cover the entirety of transcripts, only a comparatively small number of splicing informative alignments are generated for any given splice site, leading to a decreased ability to detect and/or robustly quantify many splice isoforms. To address this problem, we have recently described a method termed Multiplexed Primer Extension sequencing, or MPE-seq, which uses pools of reverse transcription primers to target sequencing to user selected loci. By targeting reverse transcription to pre-mRNA splice junctions, this approach enables a dramatic enrichment in the fraction of splicing informative alignments generated per splicing event, yielding an increase in both the precision with which splicing efficiency can be measured, and in the detection of splice isoforms including rare splicing intermediates. Here we provide a brief review of the shortcomings associated with RNA-seq that drove our development of MPE-seq, as well as a detailed protocol for implementation of MPE-seq.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interest:
The authors have no competing interests to declare.
Figures
Similar articles
-
Detection of splice isoforms and rare intermediates using multiplexed primer extension sequencing.Nat Methods. 2019 Jan;16(1):55-58. doi: 10.1038/s41592-018-0258-x. Epub 2018 Dec 20. Nat Methods. 2019. PMID: 30573814 Free PMC article.
-
Read-Split-Run: an improved bioinformatics pipeline for identification of genome-wide non-canonical spliced regions using RNA-Seq data.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):503. doi: 10.1186/s12864-016-2896-7. BMC Genomics. 2016. PMID: 27556805 Free PMC article.
-
RNA-Seq approach for accurate characterization of splicing efficiency of yeast introns.Methods. 2020 Apr 1;176:25-33. doi: 10.1016/j.ymeth.2019.03.019. Epub 2019 Mar 26. Methods. 2020. PMID: 30926533
-
Cancer-Associated Perturbations in Alternative Pre-messenger RNA Splicing.Cancer Treat Res. 2013;158:41-94. doi: 10.1007/978-3-642-31659-3_3. Cancer Treat Res. 2013. PMID: 24222354 Review.
-
Mechanisms and Regulation of Alternative Pre-mRNA Splicing.Annu Rev Biochem. 2015;84:291-323. doi: 10.1146/annurev-biochem-060614-034316. Epub 2015 Mar 12. Annu Rev Biochem. 2015. PMID: 25784052 Free PMC article. Review.
Cited by
-
The Transition from Cancer "omics" to "epi-omics" through Next- and Third-Generation Sequencing.Life (Basel). 2022 Dec 2;12(12):2010. doi: 10.3390/life12122010. Life (Basel). 2022. PMID: 36556377 Free PMC article. Review.
-
Machine learning-optimized targeted detection of alternative splicing.bioRxiv [Preprint]. 2024 Sep 24:2024.09.20.614162. doi: 10.1101/2024.09.20.614162. bioRxiv. 2024. Update in: Nucleic Acids Res. 2025 Jan 24;53(3):gkae1260. doi: 10.1093/nar/gkae1260. PMID: 39386495 Free PMC article. Updated. Preprint.
-
Cells resist starvation through a nutrient stress splice switch.Nucleic Acids Res. 2025 Jun 20;53(12):gkaf525. doi: 10.1093/nar/gkaf525. Nucleic Acids Res. 2025. PMID: 40550515 Free PMC article.
-
Machine learning-optimized targeted detection of alternative splicing.Nucleic Acids Res. 2025 Jan 24;53(3):gkae1260. doi: 10.1093/nar/gkae1260. Nucleic Acids Res. 2025. PMID: 39727154 Free PMC article.
-
Transcript-specific determinants of pre-mRNA splicing revealed through in vivo kinetic analyses of the 1st and 2nd chemical steps.Mol Cell. 2022 Aug 18;82(16):2967-2981.e6. doi: 10.1016/j.molcel.2022.06.020. Epub 2022 Jul 12. Mol Cell. 2022. PMID: 35830855 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
