

Loss of Adaptive Myelination Contributes to Methotrexate Chemotherapy-Related Cognitive Impairment
- PMID: 31122677
- PMCID: PMC6697075
- DOI: 10.1016/j.neuron.2019.04.032
Loss of Adaptive Myelination Contributes to Methotrexate Chemotherapy-Related Cognitive Impairment
Abstract
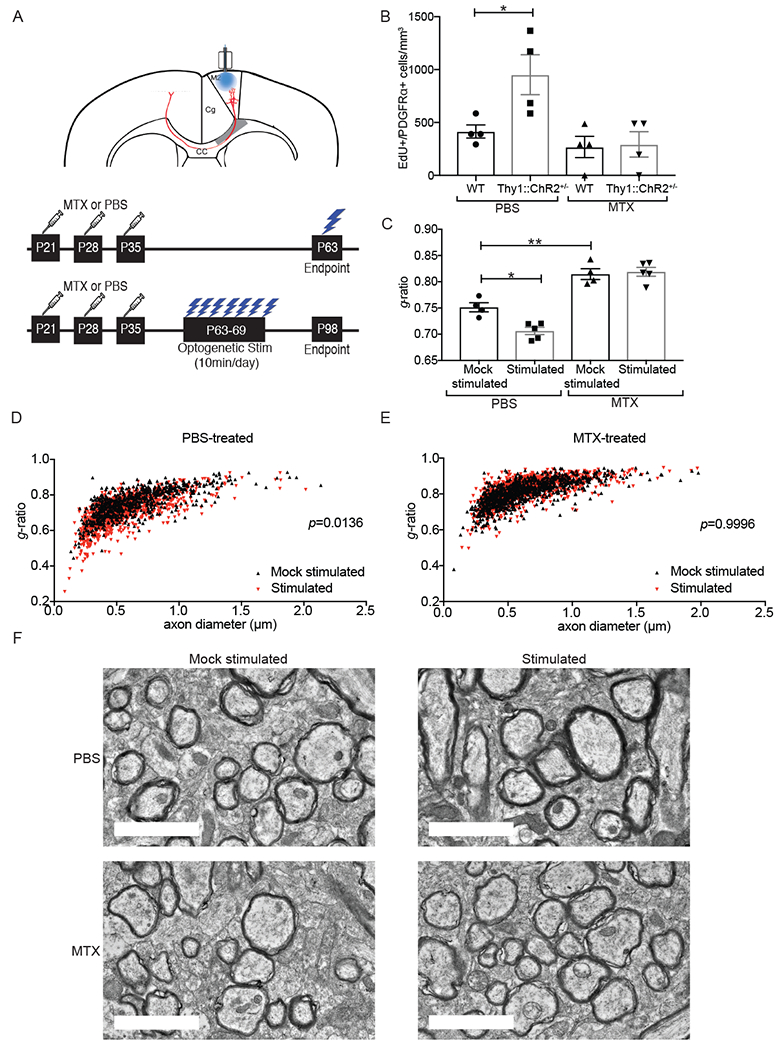
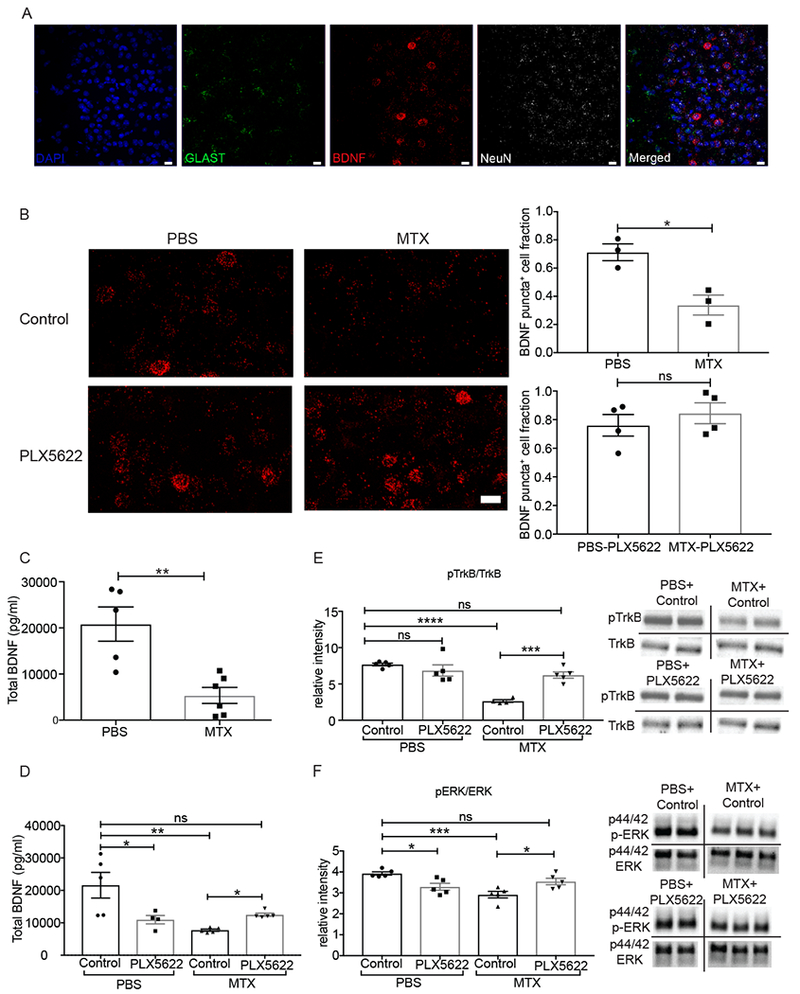
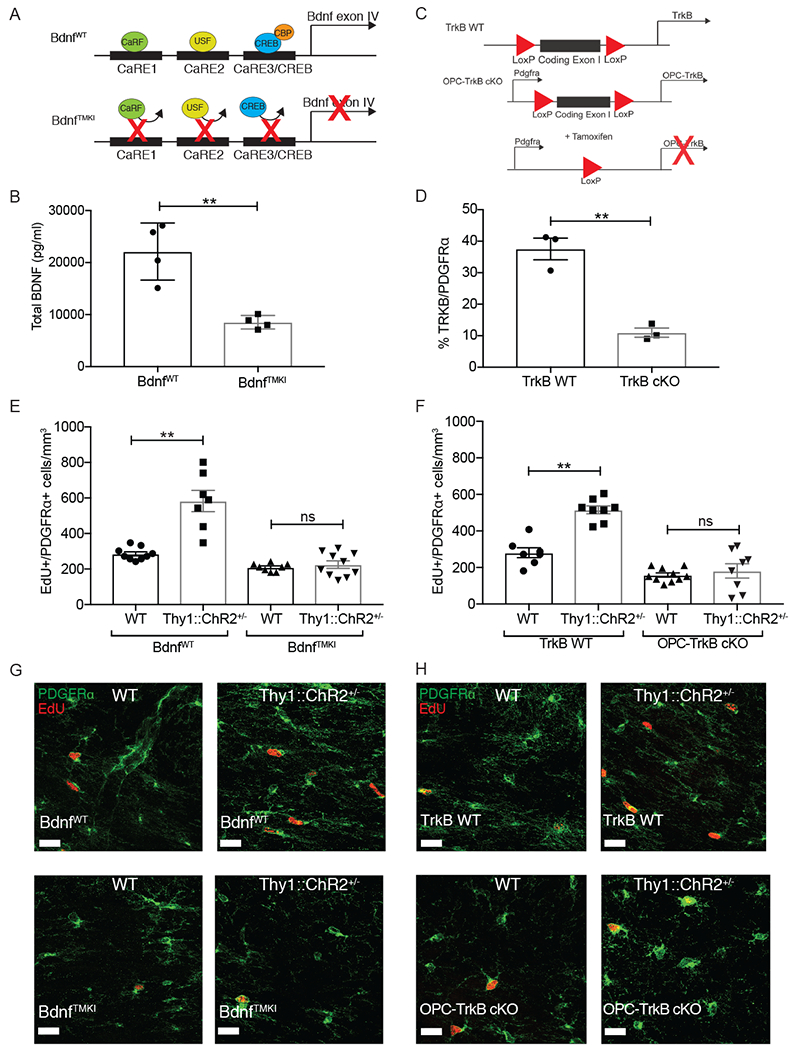
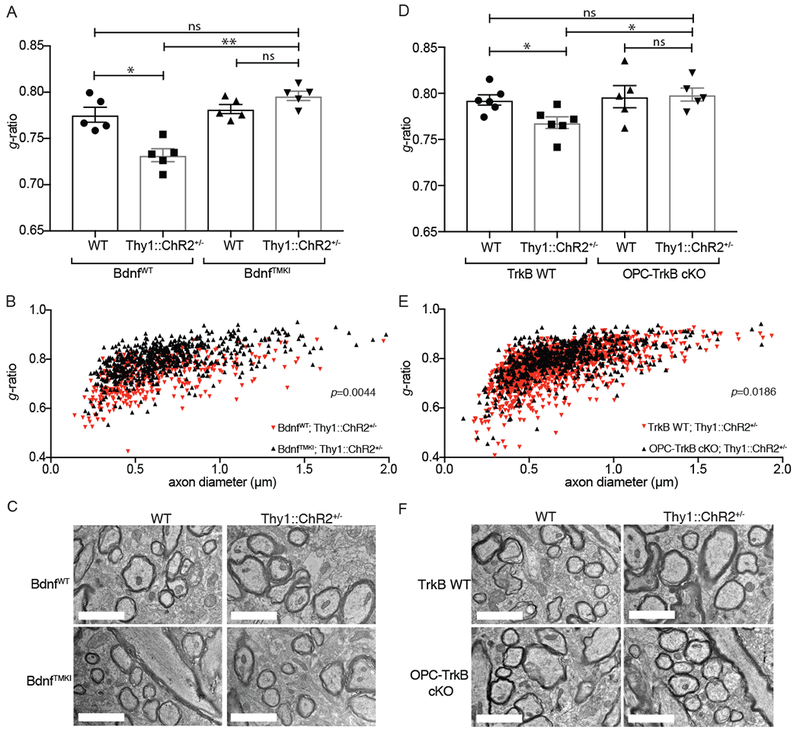
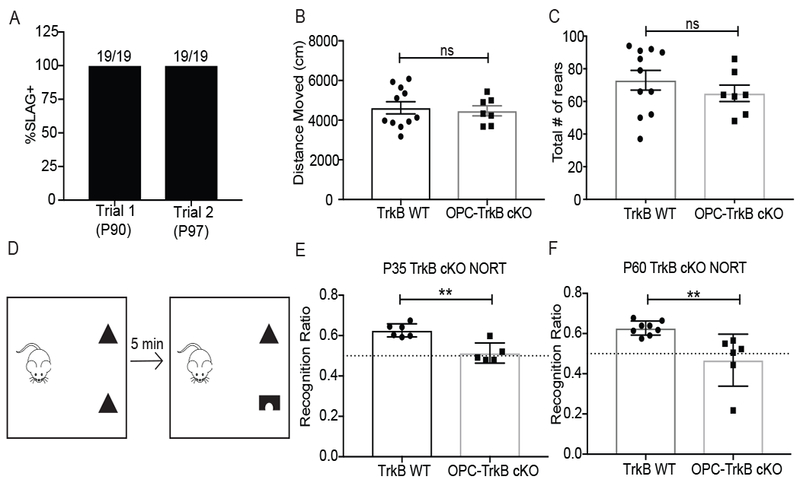
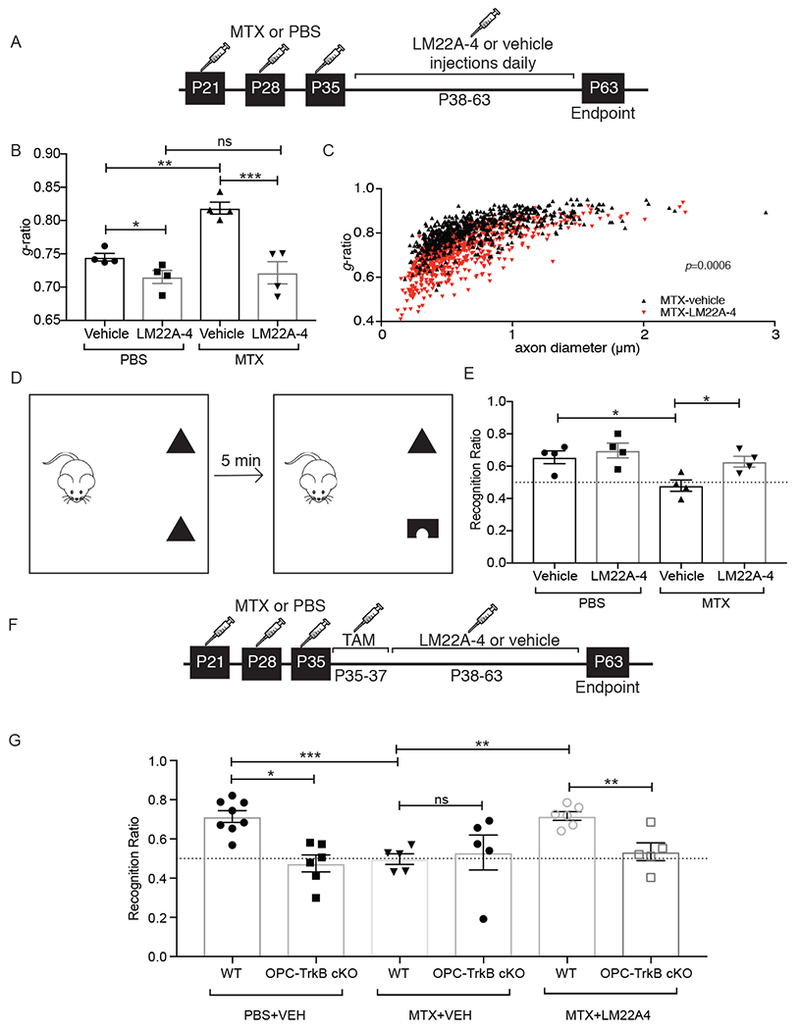
Activity-dependent myelination is thought to contribute to adaptive neurological function. However, the mechanisms by which activity regulates myelination and the extent to which myelin plasticity contributes to non-motor cognitive functions remain incompletely understood. Using a mouse model of chemotherapy-related cognitive impairment (CRCI), we recently demonstrated that methotrexate (MTX) chemotherapy induces complex glial dysfunction for which microglial activation is central. Here, we demonstrate that remote MTX exposure blocks activity-regulated myelination. MTX decreases cortical Bdnf expression, which is restored by microglial depletion. Bdnf-TrkB signaling is a required component of activity-dependent myelination. Oligodendrocyte precursor cell (OPC)-specific TrkB deletion in chemotherapy-naive mice results in impaired cognitive behavioral performance. A small-molecule TrkB agonist rescues both myelination and cognitive impairment after MTX chemotherapy. This rescue after MTX depends on intact TrkB expression in OPCs. Taken together, these findings demonstrate a molecular mechanism required for adaptive myelination that is aberrant in CRCI due to microglial activation.
Keywords: BDNF; TrkB; adaptive myelination; chemotherapy-related cognitive impairment; myelin; myelin plasticity; oligodendrocyte; oligodendrocyte precursor cell.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Comment in
-
Aberrant adaptive myelination in 'chemobrain'.Nat Rev Neurosci. 2019 Aug;20(8):448-449. doi: 10.1038/s41583-019-0194-5. Nat Rev Neurosci. 2019. PMID: 31168084 No abstract available.
References
-
- Aukema EJ, Caan MW, Oudhuis N, Majoie CB, Vos FM, Reneman L, Last BF, Grootenhuis MA, and Schouten-van Meeteren AY (2009). White matter fractional anisotropy correlates with speed of processing and motor speed in young childhood cancer survivors. Int J Radiat Oncol Biol Phys 74, 837–843. - PubMed
-
- Barres BA, and Raff MC (1993). Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
