

Binding of an X-Specific Condensin Correlates with a Reduction in Active Histone Modifications at Gene Regulatory Elements
- PMID: 31123040
- PMCID: PMC6614895
- DOI: 10.1534/genetics.119.302254
Binding of an X-Specific Condensin Correlates with a Reduction in Active Histone Modifications at Gene Regulatory Elements
Abstract
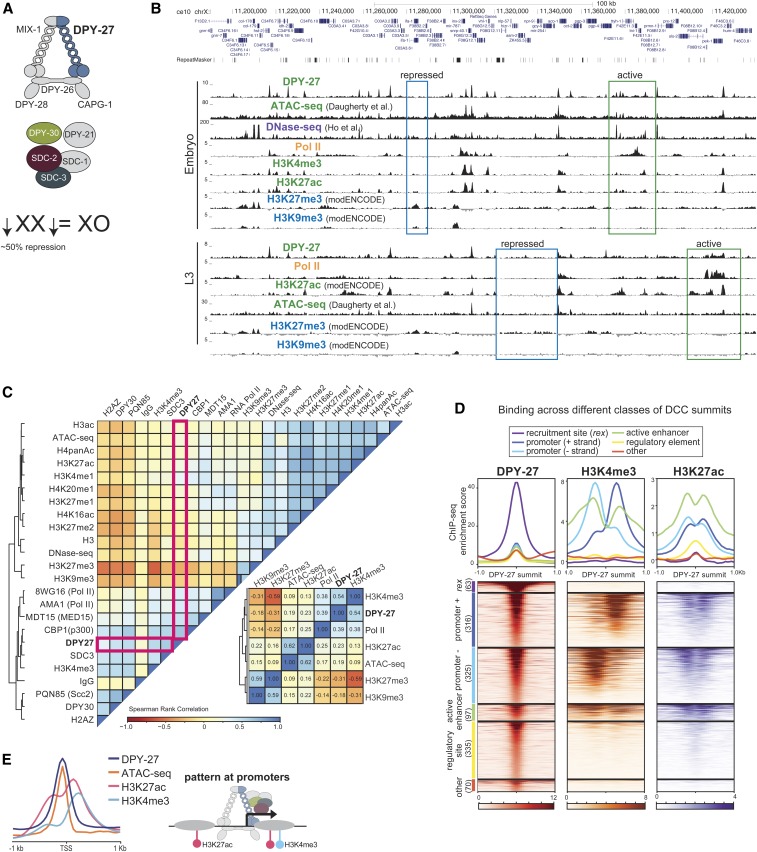
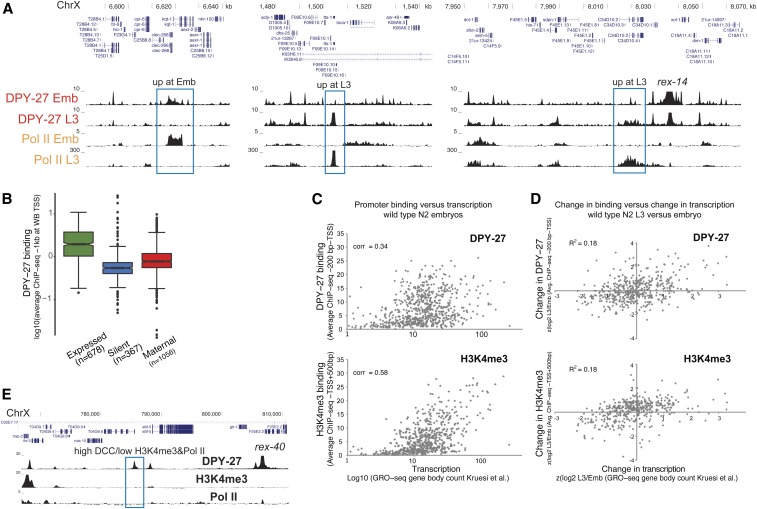
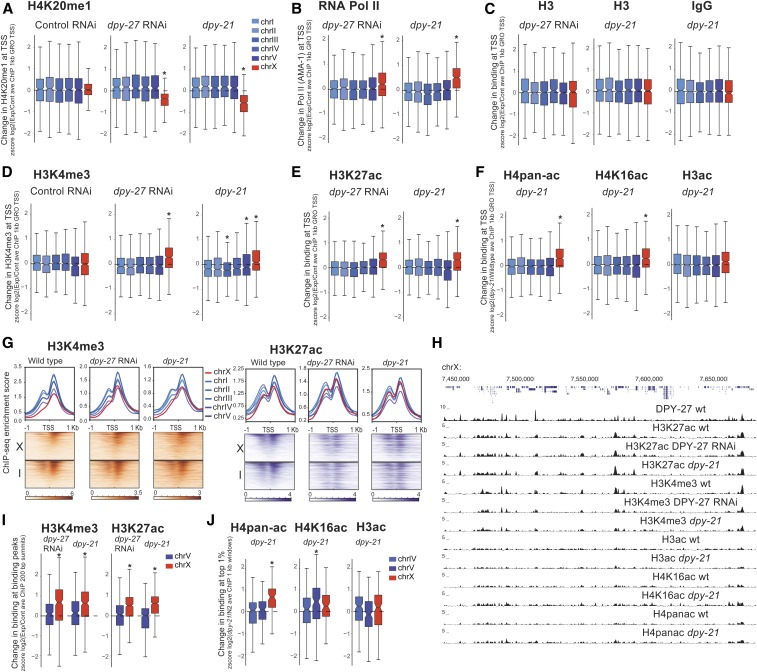
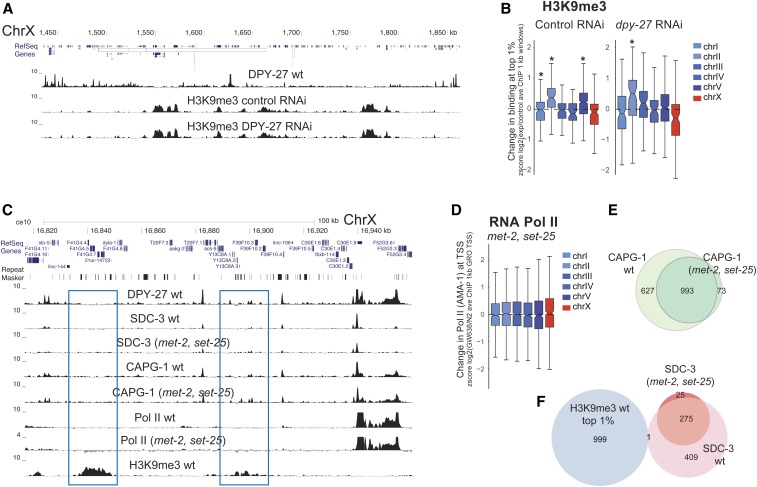
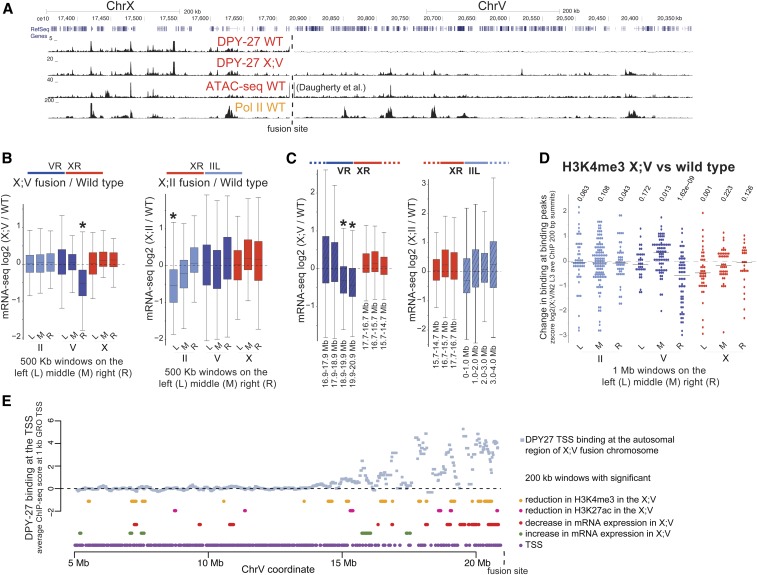
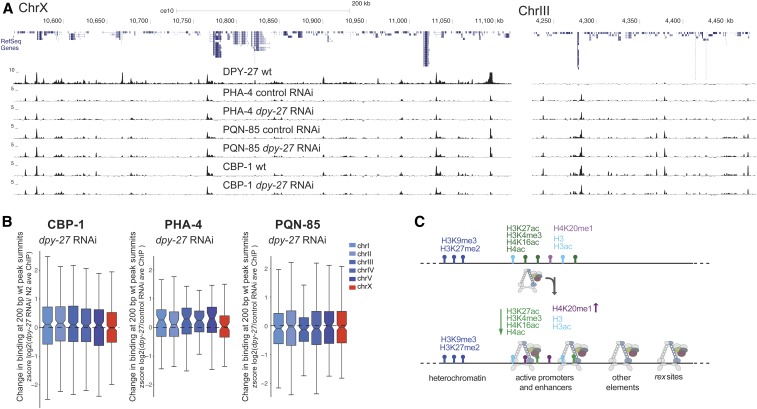
Condensins are evolutionarily conserved protein complexes that are required for chromosome segregation during cell division and genome organization during interphase. In Caenorhabditis elegans, a specialized condensin, which forms the core of the dosage compensation complex (DCC), binds to and represses X chromosome transcription. Here, we analyzed DCC localization and the effect of DCC depletion on histone modifications, transcription factor binding, and gene expression using chromatin immunoprecipitation sequencing and mRNA sequencing. Across the X, the DCC accumulates at accessible gene regulatory sites in active chromatin and not heterochromatin. The DCC is required for reducing the levels of activating histone modifications, including H3K4me3 and H3K27ac, but not repressive modification H3K9me3. In X-to-autosome fusion chromosomes, DCC spreading into the autosomal sequences locally reduces gene expression, thus establishing a direct link between DCC binding and repression. Together, our results indicate that DCC-mediated transcription repression is associated with a reduction in the activity of X chromosomal gene regulatory elements.
Keywords: C. elegans; X chromosome; chromatin; condensin; dosage compensation; gene expression; gene regulation; histone modifications; transcription.
Copyright © 2019 by the Genetics Society of America.
Figures
Similar articles
-
Condensin IDC, DPY-21, and CEC-4 maintain X chromosome repression in C. elegans.PLoS Genet. 2025 Apr 9;21(4):e1011247. doi: 10.1371/journal.pgen.1011247. eCollection 2025 Apr. PLoS Genet. 2025. PMID: 40203054 Free PMC article.
-
SUMOylation is essential for sex-specific assembly and function of the Caenorhabditis elegans dosage compensation complex on X chromosomes.Proc Natl Acad Sci U S A. 2013 Oct 1;110(40):E3810-9. doi: 10.1073/pnas.1315793110. Epub 2013 Sep 16. Proc Natl Acad Sci U S A. 2013. PMID: 24043781 Free PMC article.
-
Condensin-driven remodelling of X chromosome topology during dosage compensation.Nature. 2015 Jul 9;523(7559):240-4. doi: 10.1038/nature14450. Epub 2015 Jun 1. Nature. 2015. PMID: 26030525 Free PMC article.
-
Caenorhabditis elegans Dosage Compensation: Insights into Condensin-Mediated Gene Regulation.Trends Genet. 2018 Jan;34(1):41-53. doi: 10.1016/j.tig.2017.09.010. Epub 2017 Oct 13. Trends Genet. 2018. PMID: 29037439 Free PMC article. Review.
-
Condensin function in dosage compensation.Epigenetics. 2009 May 16;4(4):212-5. doi: 10.4161/epi.8957. Epub 2009 May 7. Epigenetics. 2009. PMID: 19483464 Review.
Cited by
-
Topoisomerases I and II facilitate condensin DC translocation to organize and repress X chromosomes in C. elegans.Mol Cell. 2022 Nov 17;82(22):4202-4217.e5. doi: 10.1016/j.molcel.2022.10.002. Epub 2022 Oct 26. Mol Cell. 2022. PMID: 36302374 Free PMC article.
-
Increased gene dosage and mRNA expression from chromosomal duplications in Caenorhabditis elegans.G3 (Bethesda). 2022 Jul 29;12(8):jkac151. doi: 10.1093/g3journal/jkac151. G3 (Bethesda). 2022. PMID: 35731207 Free PMC article.
-
Characterization of histone modification patterns and prediction of novel promoters using functional principal component analysis.PLoS One. 2020 May 27;15(5):e0233630. doi: 10.1371/journal.pone.0233630. eCollection 2020. PLoS One. 2020. PMID: 32459819 Free PMC article.
-
Revisiting Fur Regulon Leads to a Comprehensive Understanding of Iron and Fur Regulation.Int J Mol Sci. 2023 May 22;24(10):9078. doi: 10.3390/ijms24109078. Int J Mol Sci. 2023. PMID: 37240425 Free PMC article.
-
Analysis of developmental gene expression using smFISH and in silico staging of C. elegans embryos.bioRxiv [Preprint]. 2024 May 16:2024.05.15.594414. doi: 10.1101/2024.05.15.594414. bioRxiv. 2024. PMID: 38798598 Free PMC article. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
