

Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer
- PMID: 31123059
- PMCID: PMC6672049
- DOI: 10.1101/gad.326066.119
Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer
Abstract
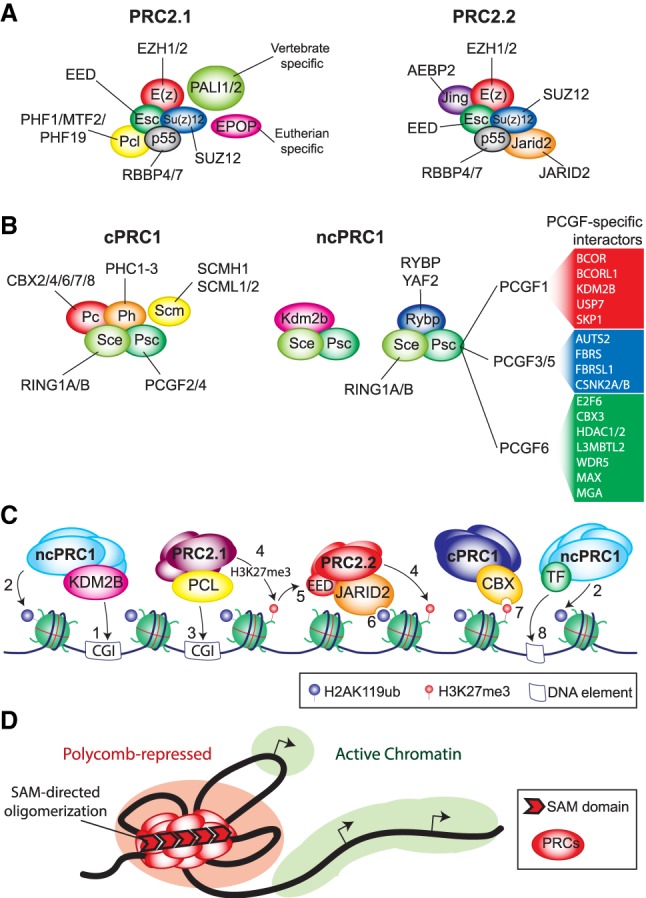
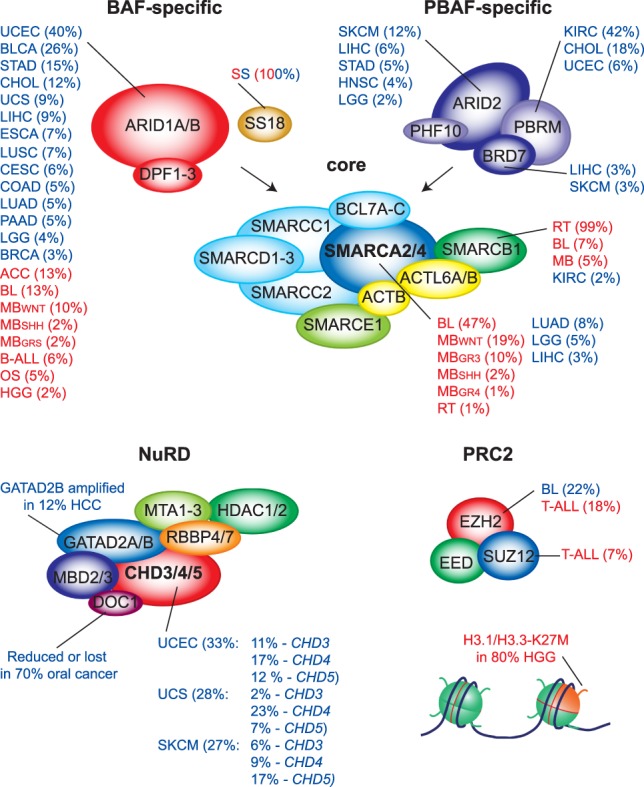
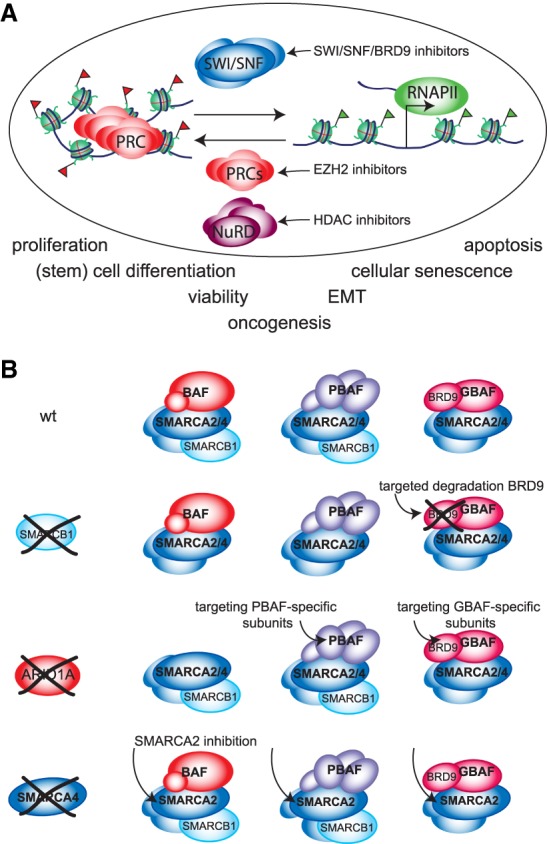
Changes in chromatin structure mediated by ATP-dependent nucleosome remodelers and histone modifying enzymes are integral to the process of gene regulation. Here, we review the roles of the SWI/SNF (switch/sucrose nonfermenting) and NuRD (nucleosome remodeling and deacetylase) and the Polycomb system in chromatin regulation and cancer. First, we discuss the basic molecular mechanism of nucleosome remodeling, and how this controls gene transcription. Next, we provide an overview of the functional organization and biochemical activities of SWI/SNF, NuRD, and Polycomb complexes. We describe how, in metazoans, the balance of these activities is central to the proper regulation of gene expression and cellular identity during development. Whereas SWI/SNF counteracts Polycomb, NuRD facilitates Polycomb repression on chromatin. Finally, we discuss how disruptions of this regulatory equilibrium contribute to oncogenesis, and how new insights into the biological functions of remodelers and Polycombs are opening avenues for therapeutic interventions on a broad range of cancer types.
Keywords: NuRD; Polycomb; SWI/SNF; cancer; chromatin.
© 2019 Bracken et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
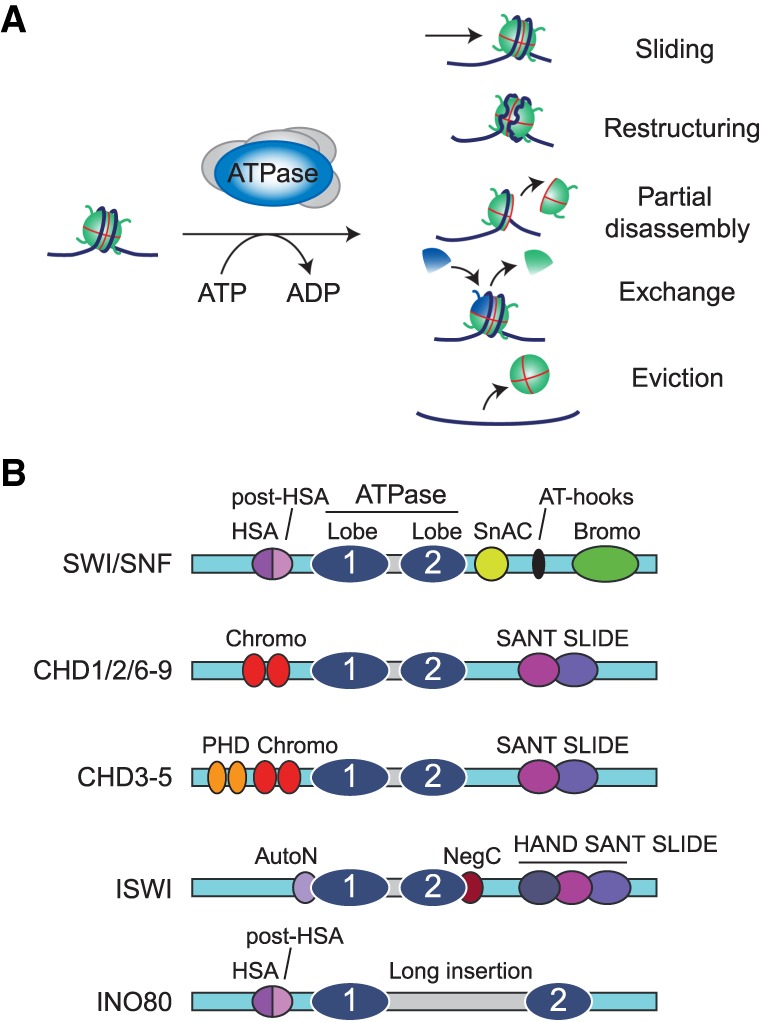
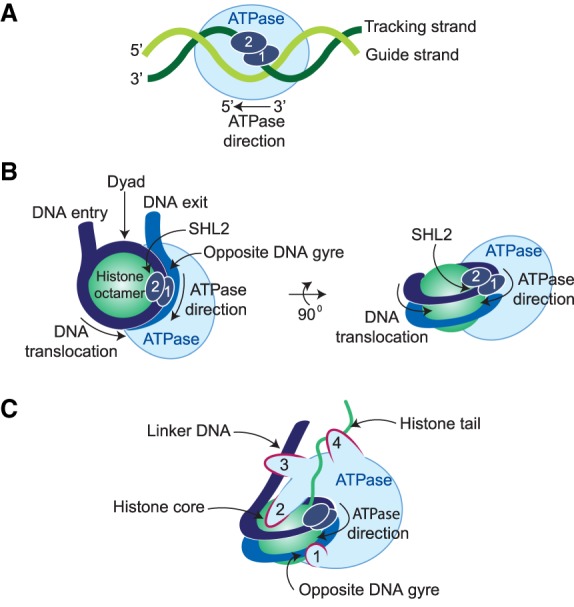
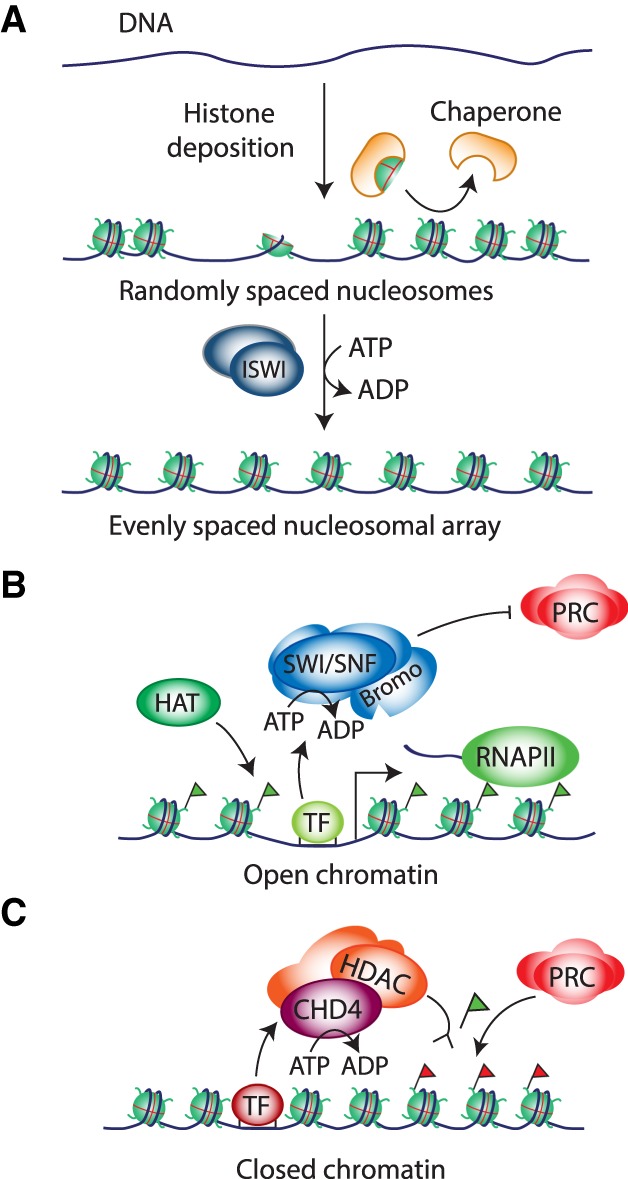
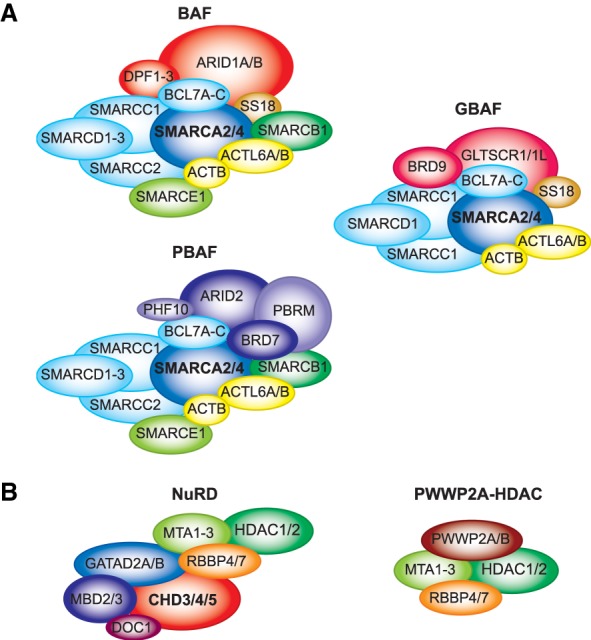
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources