

Sildenafil Reduces Neointimal Hyperplasia after Angioplasty and Inhibits Platelet Aggregation via Activation of cGMP-dependent Protein Kinase
- PMID: 31123275
- PMCID: PMC6533301
- DOI: 10.1038/s41598-019-44190-7
Sildenafil Reduces Neointimal Hyperplasia after Angioplasty and Inhibits Platelet Aggregation via Activation of cGMP-dependent Protein Kinase
Abstract
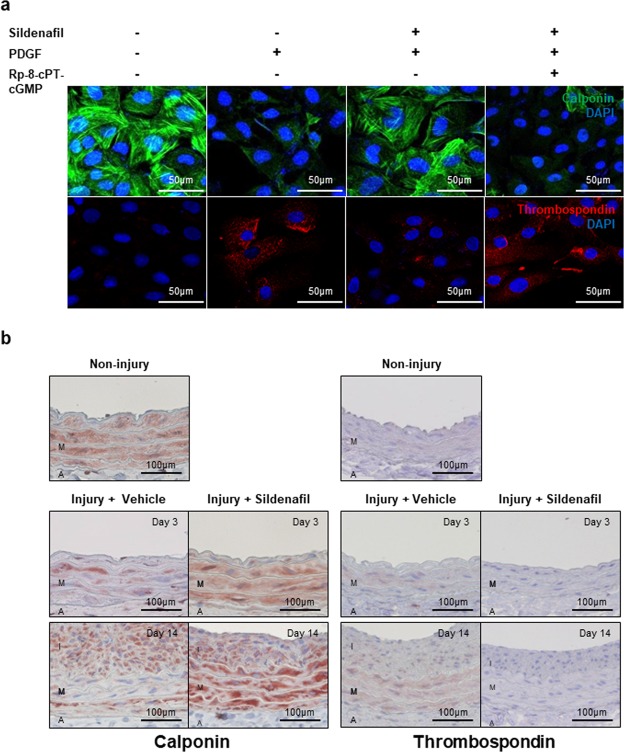
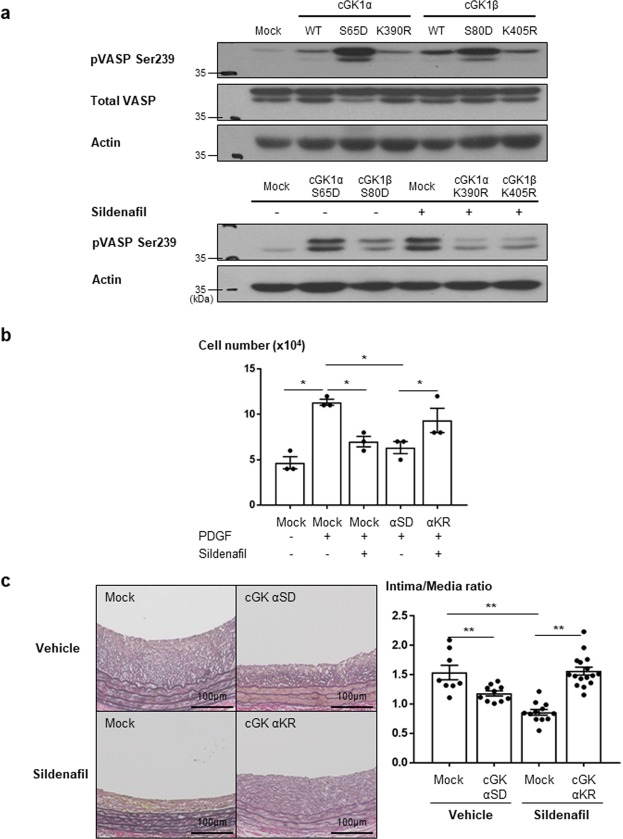
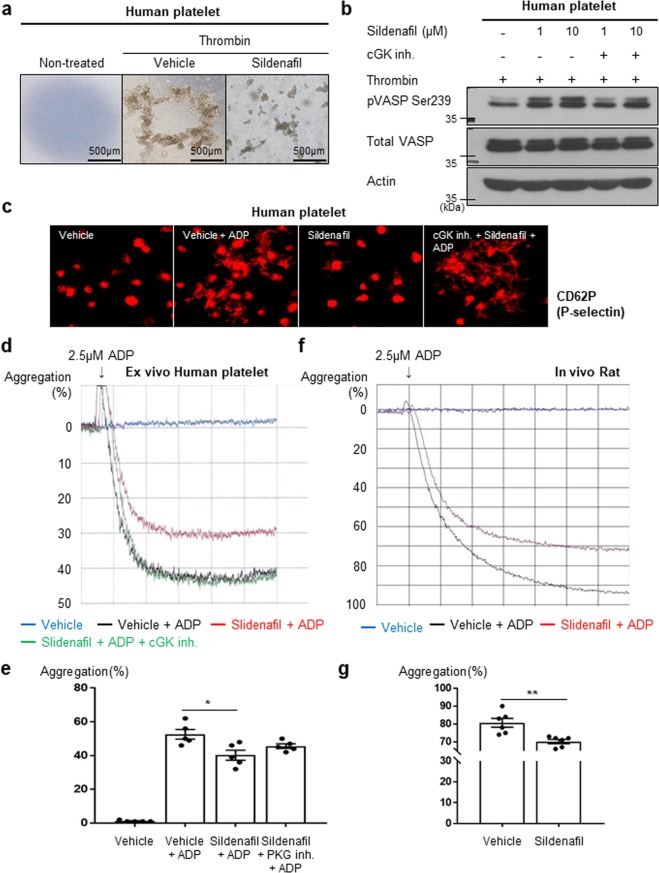
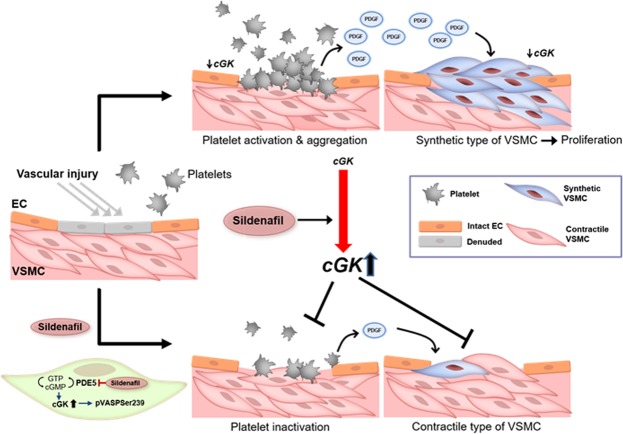
Sildenafil is known to reduce cardiac hypertrophy through cGMP-dependent protein kinase (cGK) activation. Studies have demonstrated that cGK has a central switching role in modulating vascular smooth muscle cell (VSMC) phenotype in response to vascular injury. Here, we aimed to examine the effects of cGK activation by sildenafil on neointimal formation and platelet aggregation. After vascular injury, neointimal hyperplasia in rat carotid arteries was significantly reduced in the sildenafil-treated group. This effect of sildenafil was accompanied by the reduction of viability and migration of VSMCs. Further experiments showed that the increased cGK activity by sildenafil inhibited platelet-derived growth factor-induced phenotype change of VSMCs from a contractile form to a synthetic one. Conversely, the use of cGK inhibitor or gene transfer of dominant-negative cGK reversed the effects of sildenafil, increasing viability of VSMCs and neointimal formation. Interestingly, sildenafil significantly inhibited platelet aggregation induced by ADP or thrombin. This effect was reversed by cGK inhibitor, suggesting that sildenafil inhibits platelet aggregation via cGK pathway. This study demonstrated that sildenafil inhibited neointimal formation and platelet aggregation via cGK pathway. These results suggest that sildenafil could be a promising candidate for drug-eluting stents for the prevention of both restenosis and stent thrombosis.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Activation of Protein Kinase G (PKG) Reduces Neointimal Hyperplasia, Inhibits Platelet Aggregation, and Facilitates Re-endothelialization.Sci Rep. 2016 Nov 11;6:36979. doi: 10.1038/srep36979. Sci Rep. 2016. PMID: 27833146 Free PMC article.
-
Targeting AGGF1 (angiogenic factor with G patch and FHA domains 1) for Blocking Neointimal Formation After Vascular Injury.J Am Heart Assoc. 2017 Jun 25;6(6):e005889. doi: 10.1161/JAHA.117.005889. J Am Heart Assoc. 2017. PMID: 28649088 Free PMC article.
-
PPARγ modulates vascular smooth muscle cell phenotype via a protein kinase G-dependent pathway and reduces neointimal hyperplasia after vascular injury.Exp Mol Med. 2013 Nov 29;45(11):e65. doi: 10.1038/emm.2013.112. Exp Mol Med. 2013. PMID: 24287871 Free PMC article.
-
Signalling by cGMP-dependent protein kinases.Mol Cell Biochem. 1996 Apr 12-26;157(1-2):23-30. doi: 10.1007/BF00227877. Mol Cell Biochem. 1996. PMID: 8739225 Review.
-
cGMP-dependent protein kinases in drug discovery.Drug Discov Today. 2005 May 1;10(9):627-34. doi: 10.1016/S1359-6446(05)03406-9. Drug Discov Today. 2005. PMID: 15894227 Review.
Cited by
-
Addressing male sexual and reproductive health in the wake of COVID-19 outbreak.J Endocrinol Invest. 2021 Feb;44(2):223-231. doi: 10.1007/s40618-020-01350-1. Epub 2020 Jul 13. J Endocrinol Invest. 2021. PMID: 32661947 Free PMC article. Review.
-
The potential therapeutic effect of phosphodiesterase 5 inhibitors in the acute ischemic stroke (AIS).Mol Cell Biochem. 2024 May;479(5):1267-1278. doi: 10.1007/s11010-023-04793-1. Epub 2023 Jul 3. Mol Cell Biochem. 2024. PMID: 37395897 Free PMC article. Review.
-
Sildenafil's effectiveness in the primary coronary slow flow phenomenon: a pilot randomised controlled clinical trial.Open Heart. 2024 Aug 25;11(2):e002772. doi: 10.1136/openhrt-2024-002772. Open Heart. 2024. PMID: 39214536 Free PMC article. Clinical Trial.
-
The NO/cGMP/PKG pathway in platelets: The therapeutic potential of PDE5 inhibitors in platelet disorders.J Thromb Haemost. 2022 Nov;20(11):2465-2474. doi: 10.1111/jth.15844. Epub 2022 Aug 21. J Thromb Haemost. 2022. PMID: 35950928 Free PMC article. Review.
-
Sildenafil for treating patients with COVID-19 and perfusion mismatch: a pilot randomized trial.Crit Care. 2022 Jan 3;26(1):1. doi: 10.1186/s13054-021-03885-y. Crit Care. 2022. PMID: 34980198 Free PMC article. Clinical Trial.
References
-
- Boolell M, et al. Sildenafil: an orally active type 5 cyclic GMP-specific phosphodiesterase inhibitor for the treatment of penile erectile dysfunction. Int J Impot Res. 1996;8:47–52. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
