

Review
doi: 10.1111/apm.12934.
Personalized medicine-concepts, technologies, and applications in inflammatory skin diseases
Affiliations
- PMID: 31124204
- PMCID: PMC6851586
- DOI: 10.1111/apm.12934
Item in Clipboard
Review
Personalized medicine-concepts, technologies, and applications in inflammatory skin diseases
APMIS.
2019 May.
Abstract
The current state, tools, and applications of personalized medicine with special emphasis on inflammatory skin diseases like psoriasis and atopic dermatitis are discussed. Inflammatory pathways are outlined as well as potential targets for monoclonal antibodies and small-molecule inhibitors.
Keywords: Atopic dermatitis; endotypes; immunology; inflammatory skin diseases; personalized medicine; precision medicine; psoriasis; targeted therapy.
© 2019 The Authors. APMIS published by John Wiley & Sons Ltd on behalf of Scandinavian Societies for Medical Microbiology and Pathology.
Figures
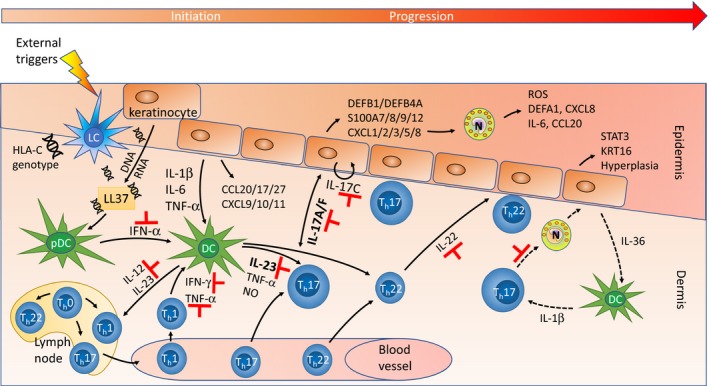
Pathways in the pathogenesis of PSO . Environmental triggers (e.g. drugs, infections, physical and psychological trauma) cause predisposed individuals to develop an autoimmune reaction, although the exact initiation mechanism is still poorly understood. One explanatory model 258 suggests that the autoantigen is LL 37 (cathelicidin antimicrobial peptide, encoded by CAMP ), which complexes with DNA and RNA released from stressed keratinocytes. This induces plasmacytoid dendritic cells (pDC s) to produce IFN ‐α, which activates dermal dendritic cells (DC s). These cells migrate to skin‐draining lymph nodes, where they secrete IL ‐12 and IL ‐23, hereby stimulating naïve T‐cells to differentiate into Th1, Th17, and Th22 cells. The Th cells are attracted into the dermis by chemokines (CCL 20, CCL 17, CCL 27, CXCL 9/10/11) released by keratinocytes. Th1 cells produce IFN ‐γ and TNF ‐α, while Th17 cells release IL ‐22 and IL ‐17 family cytokines. The latter (IL ‐17A/F) trigger epidermal keratinocytes to a feed‐forward inflammatory response 169, inducing numerous psoriasis‐associated genes [defensins, S100 proteins, chemokines; keratinocytes also produce IL ‐17 cytokines, shown is a putative, autocrine IL ‐17C loop 175] and stimulating keratinocyte proliferation. The released chemokines CXCL 1/2/3/5/8 recruit neutrophils (N), which generate ROS (reactive oxygen species), α‐defensin (DEFA 1), CXCL 8, CCL 20, and IL ‐6. IL ‐23 (released by activated DC s) stimulates differentiation and expansion of Th22 cells, which secrete IL ‐22 that induces STAT 3 and KRT 16 expression. This causes further epidermal hyperplasia and eventually formation of the psoriatic plaque. To the right (punctuated arrows) is shown the IL ‐36/IL ‐1 pathway prevalent in pustular psoriasis, which is characterized by accumulation of neutrophils; here, IL ‐17 activated neutrophils trigger increased IL ‐36 activity, which stimulates DC 's to produce IL ‐1β reinforcing the Th17 axis 179. Indicated with ⊣ are targets of approved and emerging drugs, most of which are monoclonal antibodies (see Table 2). Figure modified, mainly from van de Kerkhof & Nestle in 131, but also from Noda et al. 134, and Conrad & Gilliet 179.
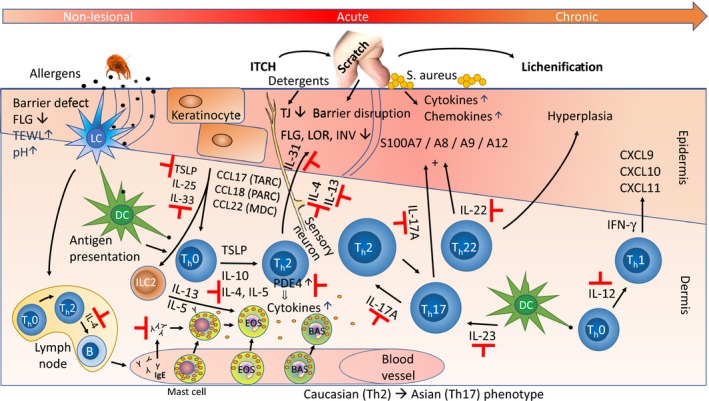
Pathways in the pathogenesis of AD . Epidermal barrier defects, which are partly due to FLG mutations, are associated with increased trans‐epidermal water loss [TEWL ), increased skin pH , and penetration of epicutaneous allergens, such as dust mite debris. When the allergens encounter antigen‐presenting epidermal Langerhans cells (LC s, for an excellent review of the interplay between LC s and the epidermis, see Clayton et al. 259] and dermal dendritic cells (DC s), this causes immune activation and recruitment of inflammatory cells, including ILC 2 [type 2 innate lymphoid cells 260] and type 2 helper T‐cells (Th2) that produce and release IL ‐4, IL ‐5, IL ‐13, and IL ‐31. These cells are considered part of the skin‐associated lymphoid tissue (SALT ), the immunologically active cutaneous microenvironment, a concept which was proposed already in 1983 by Streilein 261. IL ‐4 and IL ‐13 suppress expression of terminal differentiation genes (such as FLG , LOR , INV ), and also of tight junction (TJ ) genes 208 leading to barrier disruption, while IL ‐31 also acts directly on sensory neurons, triggering the itch–scratch cycle. This further damages the epidermis, increasing the risk of penetration by pathogens like Staphylococcus aureus. The stressed keratinocytes release TSLP , IL ‐25, and IL ‐33 that also drive Th2 differentiation. The Th2 cytokines induce IgE production in B cells and subsequently, release of inflammatory mediators (e.g. histamine) from activated (IgE bound) mast cells, basophils, and eosinophils. Th22 cells release IL ‐22, which causes epidermal hyperplasia, and also, in synergy with IL ‐17 – released from Th17 cells – induces expression of a subset of S100 family proteins. Acute AD lesions are characterized by a Th2 skewed (Th2, Th17, Th22) response, while chronic AD , which is often lichenified (thickened) by chronic scratching, progressively activates the Th1 axis with IL ‐12 release, IFN ‐γ expression and induction of chemokines (like CXCL 9/CXCL 10/CXCL 11). Indicated with ⊣ are targets of approved and emerging drugs (see Table 2 for a detailed list). Figure modified, mainly from Vakharia & Silverberg 262, based on the original by Leung 2000 263 and 2004 264. For other representations, see Noda et al. 134, Paller et al. 265, Weidinger et al. 9, Lee et al. 9, 266, and Brunner et al. 267, 268.
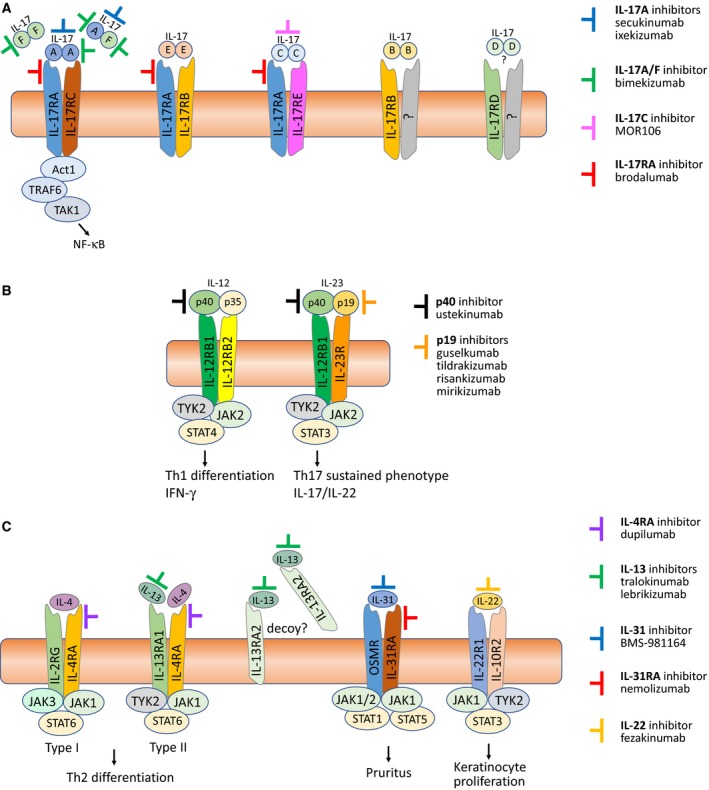
(A) Targeting the IL ‐17 family of cytokines and their receptors. The six members of the IL ‐17 cytokine family (IL ‐17A/B/C/D/E/F) are shown as functional, disulfide‐linked homodimers, as well as the IL ‐17A/F heterodimer 175. Also shown are their respective, heterodimeric receptors, each consisting of different combinations of five homologous receptor subunits (IL ‐17RA /RB /RC /RD /RE ). IL ‐17A, IL ‐17F (homodimers) and IL ‐17A/F (heterodimer) signal through the IL ‐17RA /RC receptor complex, IL ‐17E (also known as IL ‐25) via IL ‐17RA /RB , IL ‐17C via IL ‐17RA /RE , while IL ‐17B and IL ‐17D signal via yet to be determined receptors. Indicated are also monoclonal antibodies that target either the cytokines or the IL ‐17RA receptor subunit. Because IL ‐17RA is common to signaling via IL ‐17A/F/C/E/AF , blocking it will inhibit the downstream activities of all five IL ‐17 dimers. IL ‐17A/F and IL ‐17RA inhibitors have already shown substantial effect in PSO , and currently, the IL ‐17C inhibitor MOR 106 is being tested in a Phase II clinical trial in moderate to severe AD
269. (B) Targeting IL ‐12 and IL ‐23. IL ‐12 (p40/p35) and IL ‐23 (p40/p19) are heterodimers that share the same p40 subunit. IL ‐12 binds to the IL ‐12Rβ1/β2 heterodimeric receptor and stimulates JAK 2‐TYK 2 to phosphorylate mainly STAT 4, inducing IFN ‐γ and a Th1 immune response. IL ‐23 binds to the IL ‐12Rβ1/IL ‐23R heterodimeric receptor, and also induces JAK 2‐TYK 2 to phosphorylation, but primarily of STAT 3, leading to Th17 signaling and release of IL ‐17A/F and IL ‐22 270. Because the p40 subunit is common to both IL ‐12 and IL ‐23, targeting it will inhibit the effects of both cytokines 271, while the p19‐specific antagonists target only the ‘master’ regulator of Th17 development, IL ‐23 12. (C) Targeting IL ‐4/IL ‐13, IL ‐31, and IL ‐22. The two homologous cytokines, IL ‐4 and IL ‐13, drive type 2 inflammation and share many biological activities 272, the main differences being in their receptor interaction: The IL ‐4R Type I receptor consists of the IL ‐4RA and common‐gamma chain (IL ‐2RG ) subunits, and has IL ‐4 as its exclusive ligand, while the IL ‐4R Type II receptor is composed of the IL ‐4RA and IL ‐13RA 1 chains, and binds both IL ‐4 and IL ‐13. The single‐chain IL ‐13RA 2 receptor is thought to function as a decoy receptor as it seems to lack the ability to induce intracellular signaling 273. As illustrated, targeting the common IL ‐4RA subunit will inhibit the effects of both IL ‐4 and IL ‐13 signaling. IL ‐31 signals via a heterodimer consisting of IL ‐31RA and the oncostatin M receptor (OSMR ), which is also common to oncostatin M (OSM ), a member of the homologous IL ‐6 superfamily 274. The IL ‐31 receptor is found on sensory neurons in the dorsal root ganglia, where the itch sensation originates, which is why targeting IL ‐31 by e.g. nemolizumab can potentially disrupt the itch–scratch cycle of pruritic diseases like AD
275. IL ‐22 signals through the heterodimeric IL ‐22R1/IL ‐10R2 receptor and induces epidermal hyperplasia in AD , which is why the IL ‐22 antagonist fezakinumab shows some promise in treatment of severe AD
161.

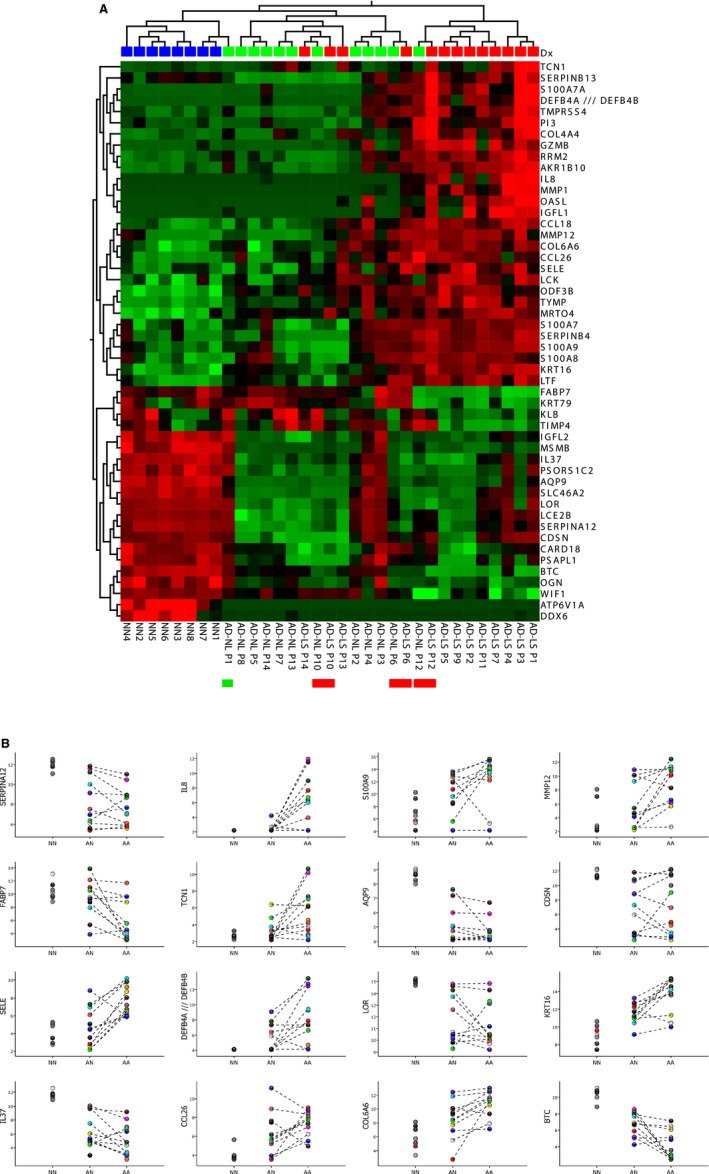
Gene expression analysis of lesional and non‐lesional skin biopsies from 13 AD patients and from eight healthy controls. (A) Heat‐map and two‐way unsupervised hierarchical clustering based on the 50 most variable genes between the three groups (non‐protein coding and orfs (open reading frames) removed). The samples cluster primarily according to disease (AD samples to the right and NN samples in the left cluster) and histology (LS to the right and NL in the middle cluster). What one also can see, is that three of the AD sample pairs (P12, P6, and P10, indicated by red bars below the heat map) cluster together, that is: there are only minor differences between the LS and NL samples from the same patient; the NL P12 sample is ‘lesional’‐like (clusters with the other LS samples), while the three ‘middle‐group’ (having overall low expression of most of the 50 DEG ) LS samples (P13, P14, P10) appear more ‘non‐lesional’ like. One NL sample (P1, indicated with a green bar below the heat‐map) clusters with the normal (NN ) group, and thus, this AD patient does not appear to have the ‘molecular scar’ typical of non‐lesional AD skin. The colors in the heat‐map signify high (red) or low (green) expression of the particular gene across samples (z‐scaled values). (B) Scatter plots of 16 selected genes, illustrating both the differences between lesional (AA ), non‐lesional (AN ), and healthy control (NN ) samples, and the variability within the groups, revealing the heterogeneity of both the diseased and ‘normal’ (healthy) population. The Y‐axis are log2‐transformed expression values (detection limit: 2–4, saturating concentrations: around 15). The samples are colored according to individual, and the dotted lines connect samples (non‐lesional and lesional) originating from the same individual. All the data used for this illustration can be accessed in GEO by its accession number, GSE 32924 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse32924 ).
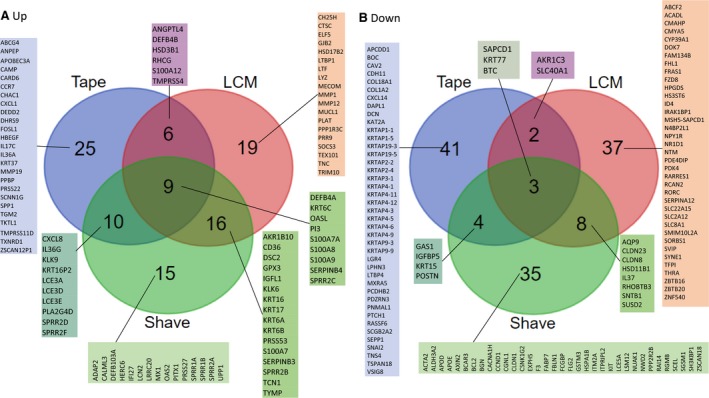
Comparison of three studies, assessing the epidermal transcriptomic profile of AD ‐LS vs AD ‐NL skin. (A) Top‐50 up DEG (LS vs NL ). (B) Top‐50 down DEG (LS vs NL ). The three studies included are Tape: Tape‐stripped skin (not in GEO ) 213; LCM : GSE 120721 222; Shave: GSE 60709 223. For the genes that are up in LS /NL epidermis, the overlap in DEG between LCM and epidermal shave is 50% (25 out of 50 genes). Fewer genes are in common between the three studies for the down DEG . For the tape‐stripping study, interestingly, many KRTAP genes appear as lower expressed in LS skin. Since KRTAP genes are associated with the hair shaft, this could suggest that there is less hair in the LS epidermis region.
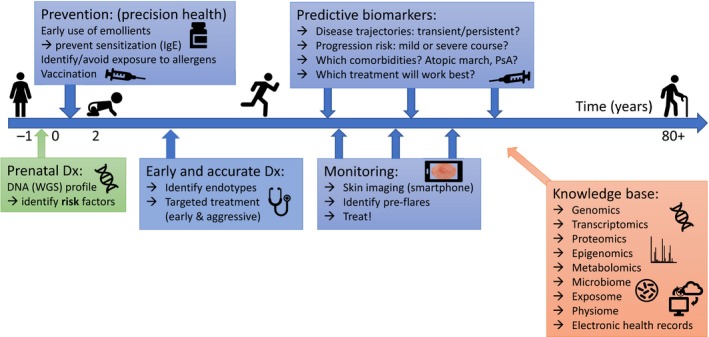
The vision of applied personalized medicine in inflammatory skin diseases. Risk factors (such as FLG mutations in AD ) can be identified before birth, enabling preventive measures (such as use of emollients) in early childhood. Identification of endotypes can guide targeted treatment, and a combination of predictive biomarkers and skin monitoring (aided by machine learning, including AI , integrating the information knowledge base) may help identify pre‐flares and optimal time and type of treatment.
Similar articles
-
Assessment of Treatment-Relevant Immune Biomarkers in Psoriasis and Atopic Dermatitis: Toward Personalized Medicine in Dermatology.J Invest Dermatol. 2023 Aug;143(8):1412-1422. doi: 10.1016/j.jid.2023.04.005. Epub 2023 Jun 20. J Invest Dermatol. 2023. PMID: 37341663 Free PMC article. Review.
-
Multi-Omics Approach to Improved Diagnosis and Treatment of Atopic Dermatitis and Psoriasis.Int J Mol Sci. 2024 Jan 15;25(2):1042. doi: 10.3390/ijms25021042. Int J Mol Sci. 2024. PMID: 38256115 Free PMC article. Review.
-
Toward Precision Medicine in Atopic Dermatitis Using Molecular-Based Approaches.Actas Dermosifiliogr. 2024 Jan;115(1):66-75. doi: 10.1016/j.ad.2023.08.003. Epub 2023 Aug 29. Actas Dermosifiliogr. 2024. PMID: 37652096 Review. English, Spanish.
-
Atopic dermatitis endotypes: knowledge for personalized medicine.Curr Opin Allergy Clin Immunol. 2022 Jun 1;22(3):153-159. doi: 10.1097/ACI.0000000000000820. Epub 2022 Feb 11. Curr Opin Allergy Clin Immunol. 2022. PMID: 35152229 Review.
-
Atopic dermatitis stratification: current and future perspective on skin and blood transcriptomic and proteomic profiling.Expert Rev Clin Immunol. 2024 Sep;20(9):1083-1088. doi: 10.1080/1744666X.2024.2323964. Epub 2024 Mar 4. Expert Rev Clin Immunol. 2024. PMID: 38436065 Review.
Cited by
-
The Translational Dermatology Initiative: Aiming at a New Disease Classification of Inflammatory Skin Diseases.JID Innov. 2025 May 13;5(5):100381. doi: 10.1016/j.xjidi.2025.100381. eCollection 2025 Sep. JID Innov. 2025. PMID: 40535547 Free PMC article.
-
Next-Generation Oral Delivery Systems: Phytosomal Hinokitiol Tablets via REGEMAT 3D Bioprinter-Based 3D Printing for Enhanced Bioavailability.Scientifica (Cairo). 2025 Jun 30;2025:6678786. doi: 10.1155/sci5/6678786. eCollection 2025. Scientifica (Cairo). 2025. PMID: 40661180 Free PMC article.
-
Review of Personalized Medicine and Pharmacogenomics of Anti-Cancer Compounds and Natural Products.Genes (Basel). 2024 Apr 8;15(4):468. doi: 10.3390/genes15040468. Genes (Basel). 2024. PMID: 38674402 Free PMC article. Review.
-
Overview of omics biomarkers in pituitary neuroendocrine tumors to design future diagnosis and treatment strategies.EPMA J. 2021 Jun 26;12(3):383-401. doi: 10.1007/s13167-021-00246-1. eCollection 2021 Sep. EPMA J. 2021. PMID: 34567287 Free PMC article. Review.
-
Omics-Driven Biomarkers of Psoriasis: Recent Insights, Current Challenges, and Future Prospects.Clin Cosmet Investig Dermatol. 2020 Aug 25;13:611-625. doi: 10.2147/CCID.S227896. eCollection 2020. Clin Cosmet Investig Dermatol. 2020. PMID: 32922059 Free PMC article. Review.
References
-
- Vail J. Pharmacogenomics: the end of trial‐and‐error medicine? Int J Pharm Compd 2007;11:59–65. - PubMed
-
- Morgan P, Brown DG, Lennard S, Anderton MJ, Barrett JC, Eriksson U, et al. Impact of a five‐dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov 2018;17:167–81. - PubMed
-
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med 2005;353:1711–23. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
