

Corrupted DNA-binding specificity and ectopic transcription underpin dominant neomorphic mutations in KLF/SP transcription factors
- PMID: 31126231
- PMCID: PMC6534859
- DOI: 10.1186/s12864-019-5805-z
Corrupted DNA-binding specificity and ectopic transcription underpin dominant neomorphic mutations in KLF/SP transcription factors
Abstract
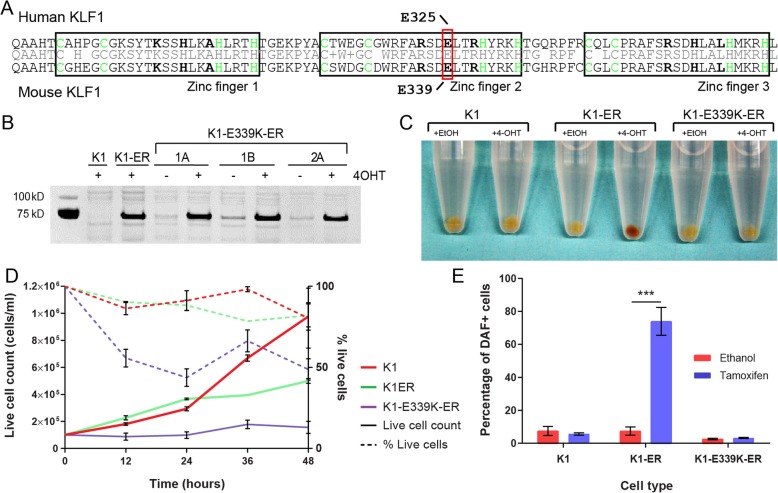
Background: Mutations in the transcription factor, KLF1, are common within certain populations of the world. Heterozygous missense mutations in KLF1 mostly lead to benign phenotypes, but a heterozygous mutation in a DNA-binding residue (E325K in human) results in severe Congenital Dyserythropoietic Anemia type IV (CDA IV); i.e. an autosomal-dominant disorder characterized by neonatal hemolysis.
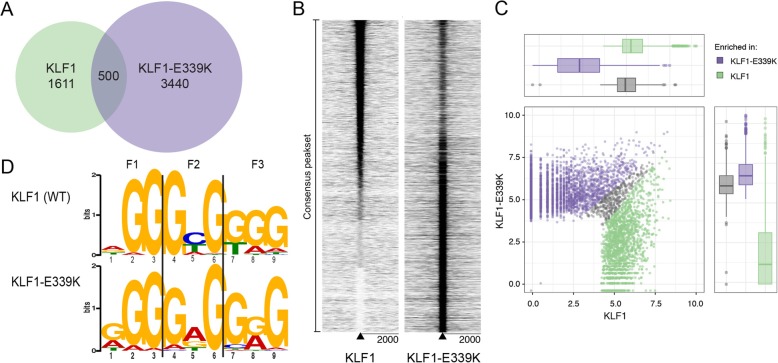
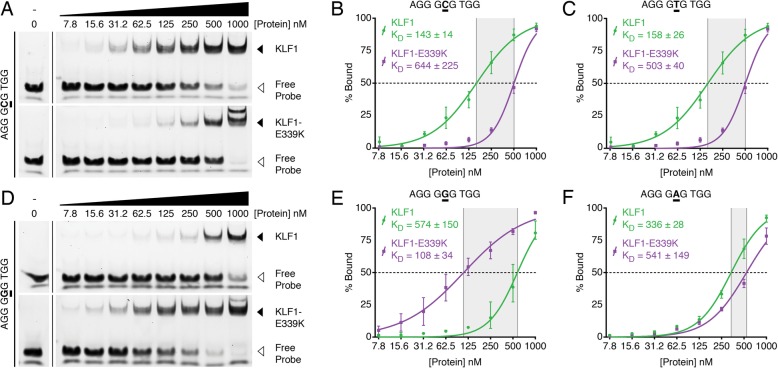
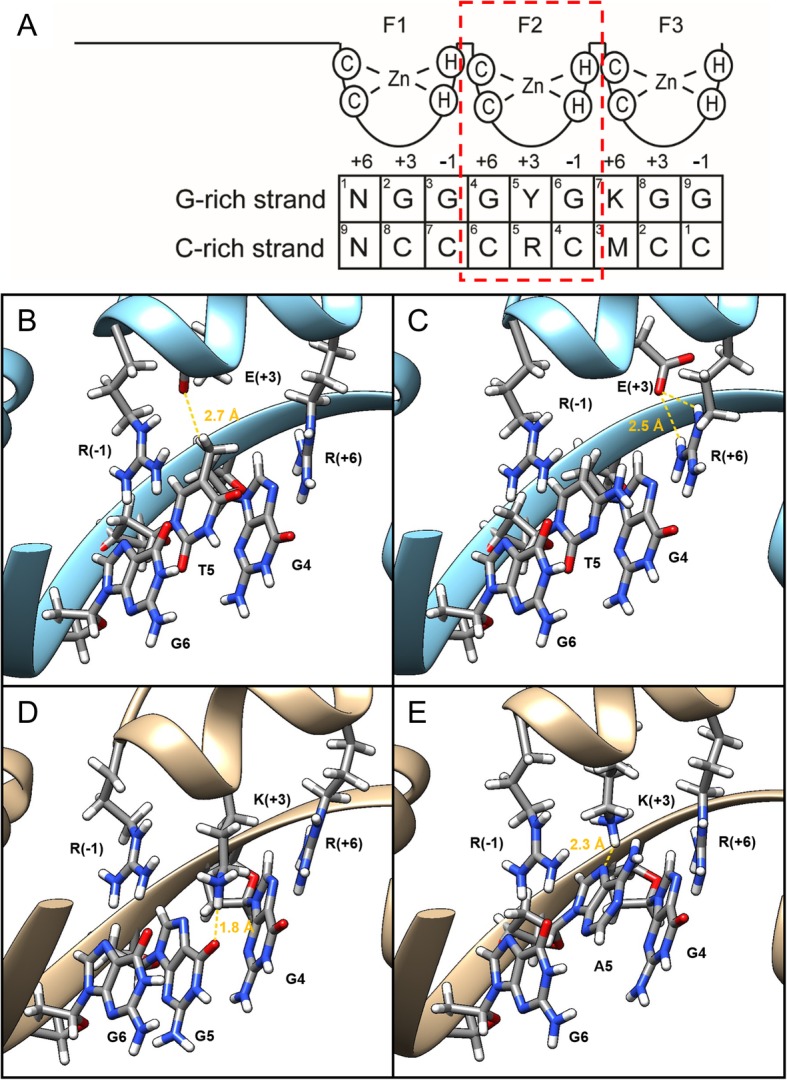
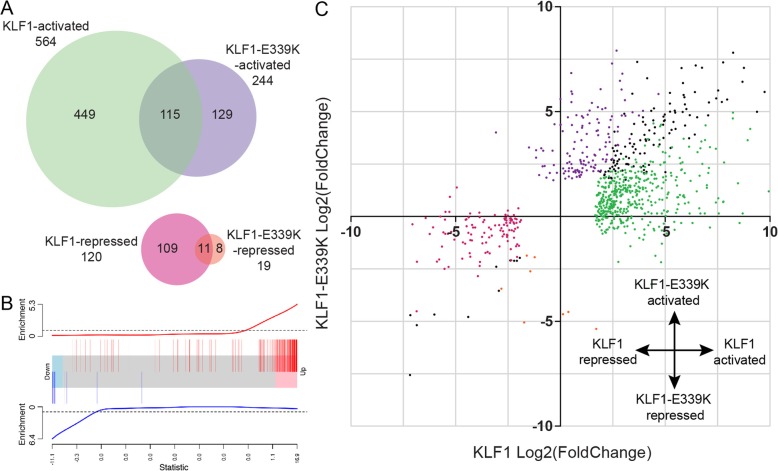
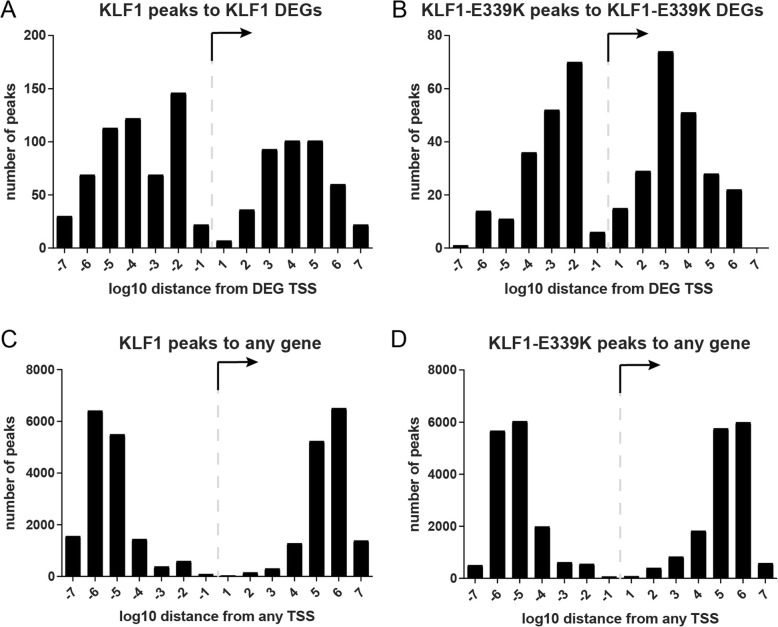
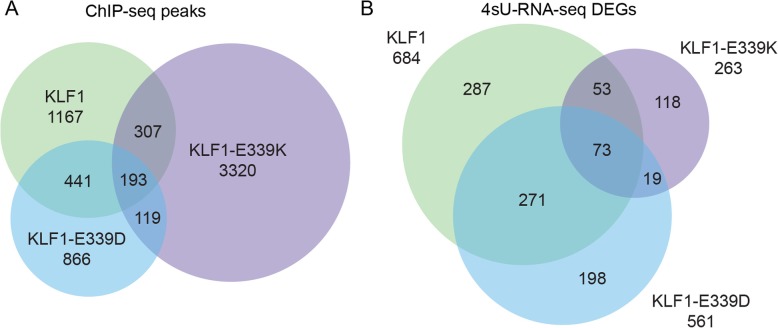
Results: To investigate the biochemical and genetic mechanism of CDA IV, we generated murine erythroid cell lines that harbor tamoxifen-inducible (ER™) versions of wild type and mutant KLF1 on a Klf1-/- genetic background. Nuclear translocation of wild type KLF1 results in terminal erythroid differentiation, whereas mutant KLF1 results in hemolysis without differentiation. The E to K variant binds poorly to the canonical 9 bp recognition motif (NGG-GYG-KGG) genome-wide but binds at high affinity to a corrupted motif (NGG-GRG-KGG). We confirmed altered DNA-binding specificity by quantitative in vitro binding assays of recombinant zinc-finger domains. Our results are consistent with previously reported structural data of KLF-DNA interactions. We employed 4sU-RNA-seq to show that a corrupted transcriptome is a direct consequence of aberrant DNA binding.
Conclusions: Since all KLF/SP family proteins bind DNA in an identical fashion, these results are likely to be generally applicable to mutations in all family members. Importantly, they explain how certain mutations in the DNA-binding domain of transcription factors can generate neomorphic functions that result in autosomal dominant disease.
Keywords: 4sU-RNA labeling; CDA; ChIP-seq; Hemolysis; KLF1.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Severe anemia caused by dominant mutations in Krüppel-like factor 1 (KLF1).Mutat Res Rev Mutat Res. 2020 Oct-Dec;786:108336. doi: 10.1016/j.mrrev.2020.108336. Epub 2020 Oct 9. Mutat Res Rev Mutat Res. 2020. PMID: 33339573 Free PMC article. Review.
-
A Krüppel-like factor 1 (KLF1) Mutation Associated with Severe Congenital Dyserythropoietic Anemia Alters Its DNA-Binding Specificity.Mol Cell Biol. 2020 Feb 12;40(5):e00444-19. doi: 10.1128/MCB.00444-19. Print 2020 Feb 12. Mol Cell Biol. 2020. PMID: 31818881 Free PMC article.
-
KLF1 mutation E325K induces cell cycle arrest in erythroid cells differentiated from congenital dyserythropoietic anemia patient-specific induced pluripotent stem cells.Exp Hematol. 2019 May;73:25-37.e8. doi: 10.1016/j.exphem.2019.03.001. Epub 2019 Mar 12. Exp Hematol. 2019. PMID: 30876823 Clinical Trial.
-
A Case of Congenital Dyserythropoeitic Anemia Type IV Caused by E325K Mutation in Erythroid Transcription Factor KLF1.J Pediatr Hematol Oncol. 2018 Aug;40(6):e389-e391. doi: 10.1097/MPH.0000000000001042. J Pediatr Hematol Oncol. 2018. PMID: 29200155
-
Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm.Blood Cells Mol Dis. 2013 Aug;51(2):71-5. doi: 10.1016/j.bcmd.2013.02.006. Epub 2013 Mar 20. Blood Cells Mol Dis. 2013. PMID: 23522491 Free PMC article. Review.
Cited by
-
A novel human cellular model of CDA IV enables comprehensive analysis revealing the molecular basis of the disease phenotype.Blood. 2023 Jun 22;141(25):3039-3054. doi: 10.1182/blood.2022018735. Blood. 2023. PMID: 37084386 Free PMC article.
-
KLF feedback loops in innate immunity.Front Immunol. 2025 Jun 4;16:1606277. doi: 10.3389/fimmu.2025.1606277. eCollection 2025. Front Immunol. 2025. PMID: 40534851 Free PMC article. Review.
-
Mutations in linker-2 of KLF1 impair expression of membrane transporters and cytoskeletal proteins causing hemolysis.Nat Commun. 2024 Aug 15;15(1):7019. doi: 10.1038/s41467-024-50579-4. Nat Commun. 2024. PMID: 39147774 Free PMC article.
-
Neural network modeling of differential binding between wild-type and mutant CTCF reveals putative binding preferences for zinc fingers 1-2.BMC Genomics. 2022 Apr 12;23(1):295. doi: 10.1186/s12864-022-08486-9. BMC Genomics. 2022. PMID: 35410161 Free PMC article.
-
Severe anemia caused by dominant mutations in Krüppel-like factor 1 (KLF1).Mutat Res Rev Mutat Res. 2020 Oct-Dec;786:108336. doi: 10.1016/j.mrrev.2020.108336. Epub 2020 Oct 9. Mutat Res Rev Mutat Res. 2020. PMID: 33339573 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
