

Horizontal Gene Transfer as an Indispensable Driver for Evolution of Neocallimastigomycota into a Distinct Gut-Dwelling Fungal Lineage
- PMID: 31126947
- PMCID: PMC6643240
- DOI: 10.1128/AEM.00988-19
Horizontal Gene Transfer as an Indispensable Driver for Evolution of Neocallimastigomycota into a Distinct Gut-Dwelling Fungal Lineage
Abstract
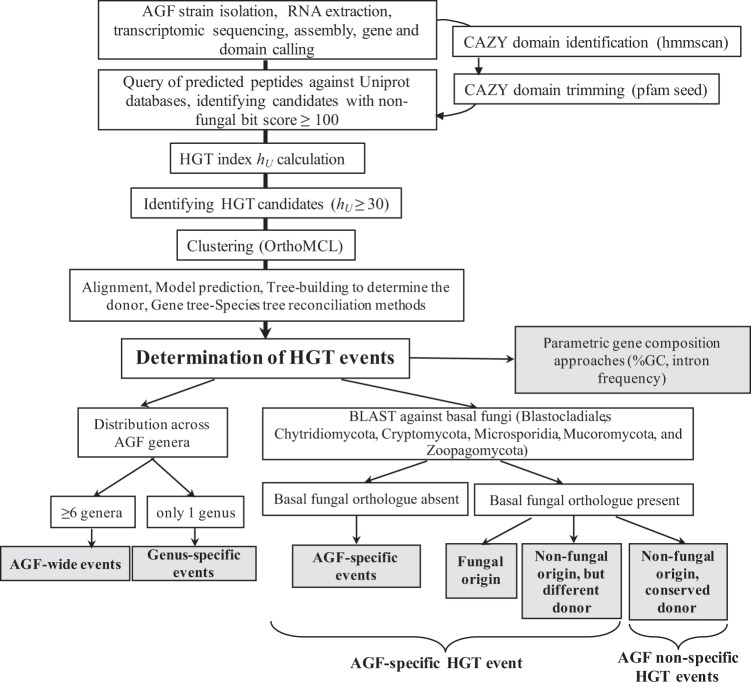
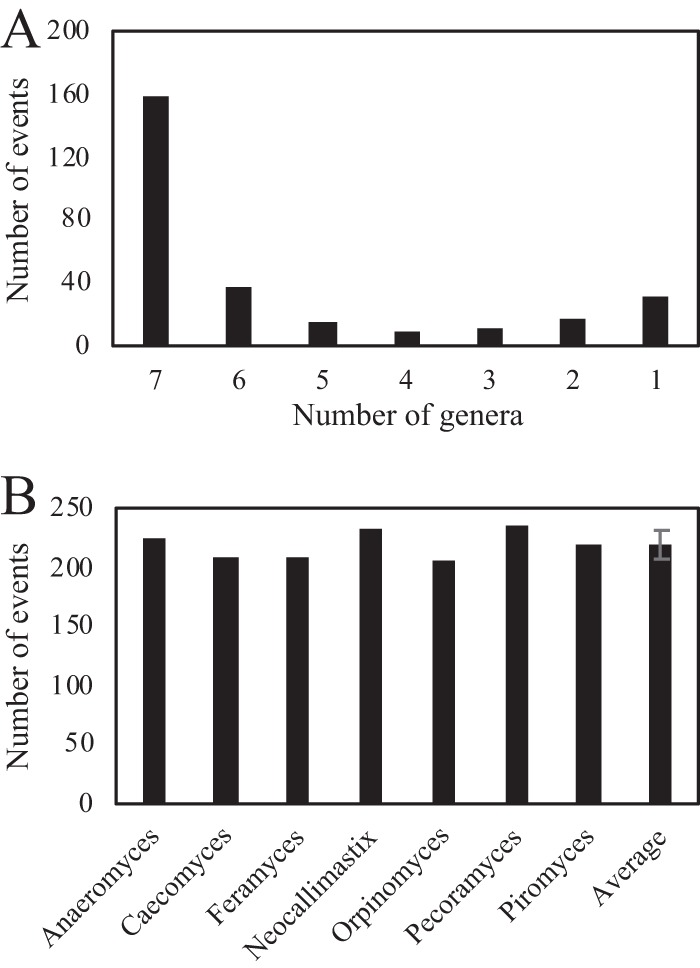
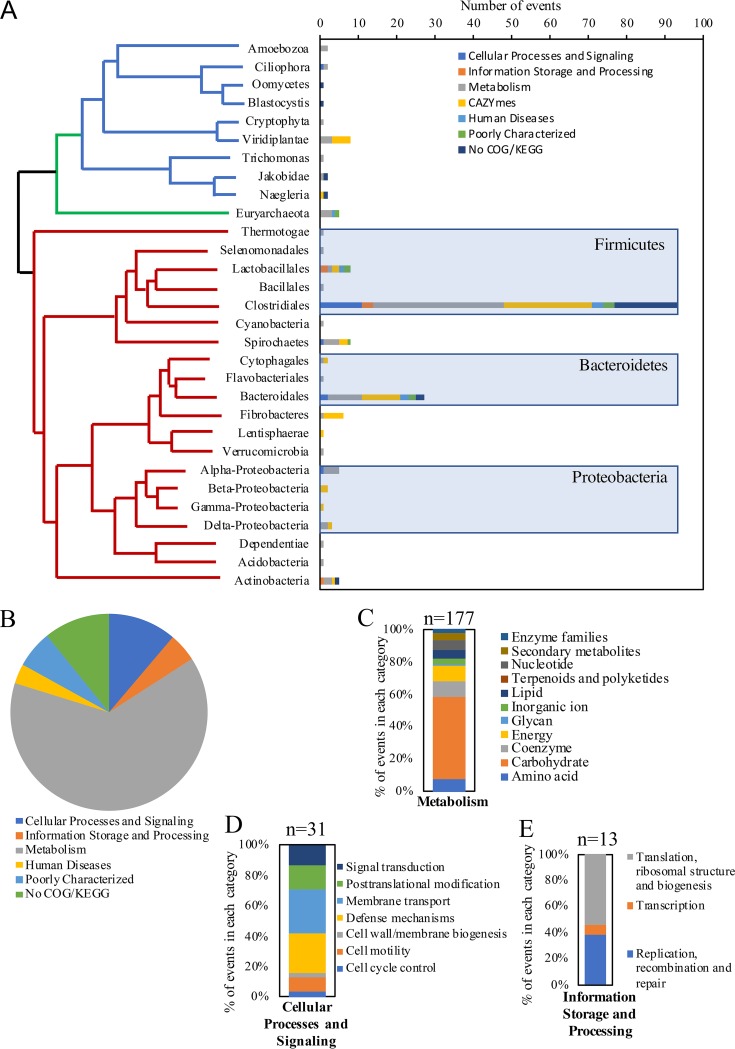
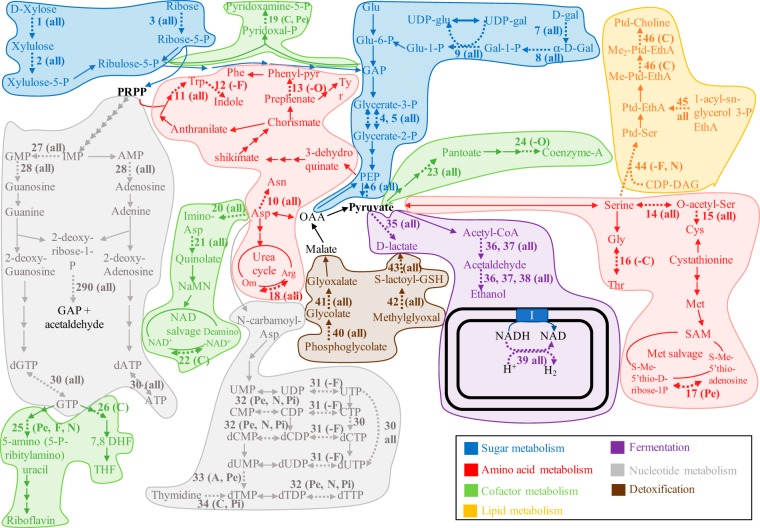
Survival and growth of the anaerobic gut fungi (AGF; Neocallimastigomycota) in the herbivorous gut necessitate the possession of multiple abilities absent in other fungal lineages. We hypothesized that horizontal gene transfer (HGT) was instrumental in forging the evolution of AGF into a phylogenetically distinct gut-dwelling fungal lineage. The patterns of HGT were evaluated in the transcriptomes of 27 AGF strains, 22 of which were isolated and sequenced in this study, and 4 AGF genomes broadly covering the breadth of AGF diversity. We identified 277 distinct incidents of HGT in AGF transcriptomes, with subsequent gene duplication resulting in an HGT frequency of 2 to 3.5% in AGF genomes. The majority of HGT events were AGF specific (91.7%) and wide (70.8%), indicating their occurrence at early stages of AGF evolution. The acquired genes allowed AGF to expand their substrate utilization range, provided new venues for electron disposal, augmented their biosynthetic capabilities, and facilitated their adaptation to anaerobiosis. The majority of donors were anaerobic fermentative bacteria prevalent in the herbivorous gut. This study strongly indicates that HGT indispensably forged the evolution of AGF as a distinct fungal phylum and provides a unique example of the role of HGT in shaping the evolution of a high-rank taxonomic eukaryotic lineage.IMPORTANCE The anaerobic gut fungi (AGF) represent a distinct basal phylum lineage (Neocallimastigomycota) commonly encountered in the rumen and alimentary tracts of herbivores. Survival and growth of anaerobic gut fungi in these anaerobic, eutrophic, and prokaryote-dominated habitats necessitates the acquisition of several traits absent in other fungal lineages. We assess here the role of horizontal gene transfer as a relatively fast mechanism for trait acquisition by the Neocallimastigomycota postsequestration in the herbivorous gut. Analysis of 27 transcriptomes that represent the broad diversity of Neocallimastigomycota identified 277 distinct HGT events, with subsequent gene duplication resulting in an HGT frequency of 2 to 3.5% in AGF genomes. These HGT events have allowed AGF to survive in the herbivorous gut by expanding their substrate utilization range, augmenting their biosynthetic pathway, providing new routes for electron disposal by expanding fermentative capacities, and facilitating their adaptation to anaerobiosis. HGT in the AGF is also shown to be mainly a cross-kingdom affair, with the majority of donors belonging to the bacteria. This study represents a unique example of the role of HGT in shaping the evolution of a high-rank taxonomic eukaryotic lineage.
Keywords: Neocallimastigomycota; anaerobic gut fungi; horizontal gene transfer.
Copyright © 2019 American Society for Microbiology.
Figures
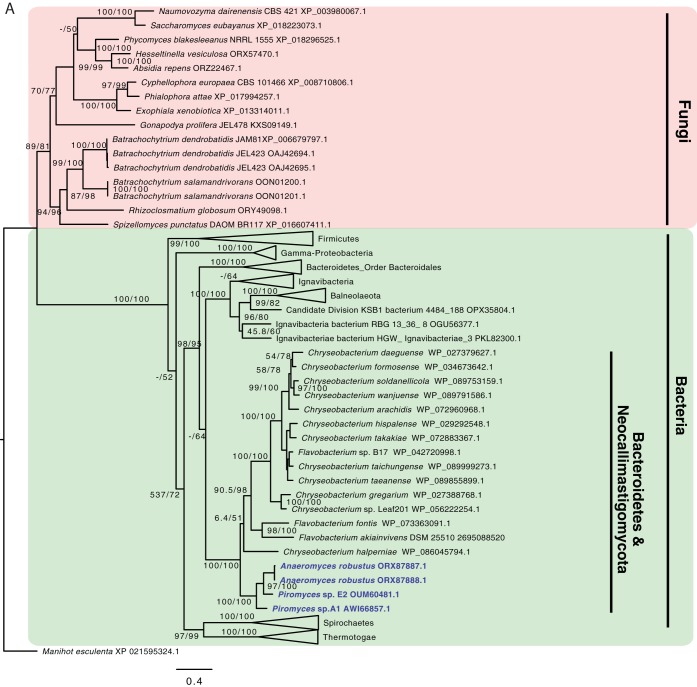
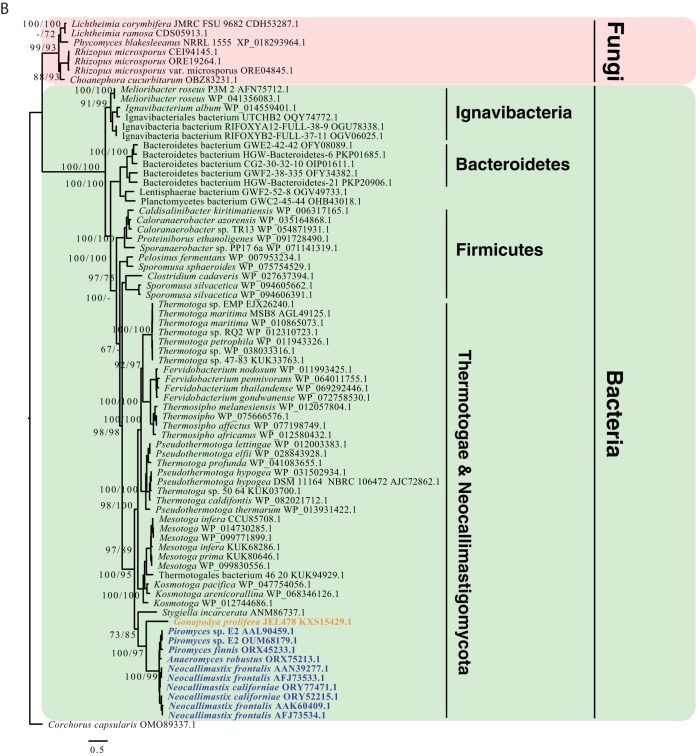
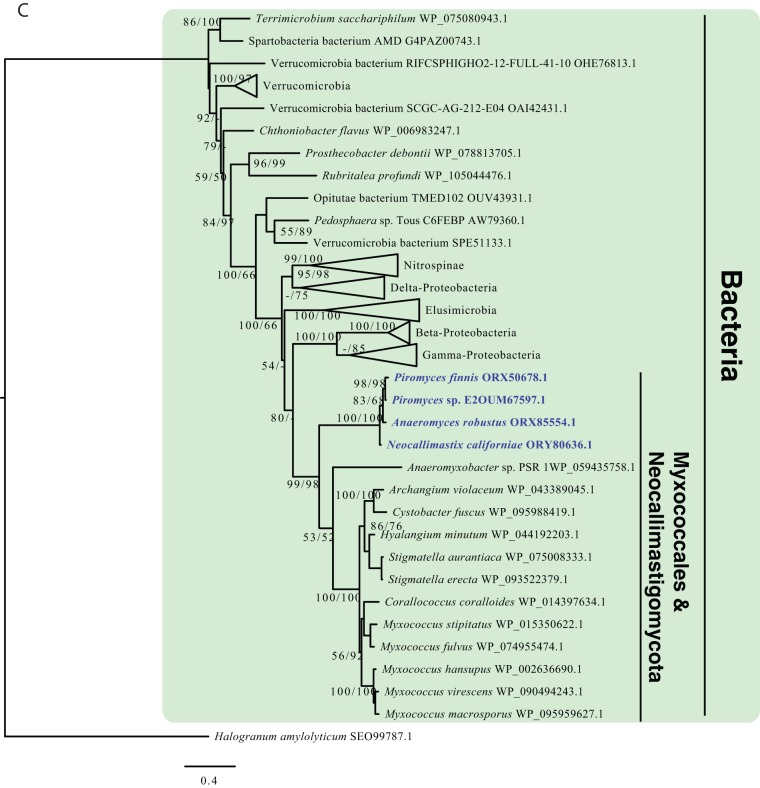
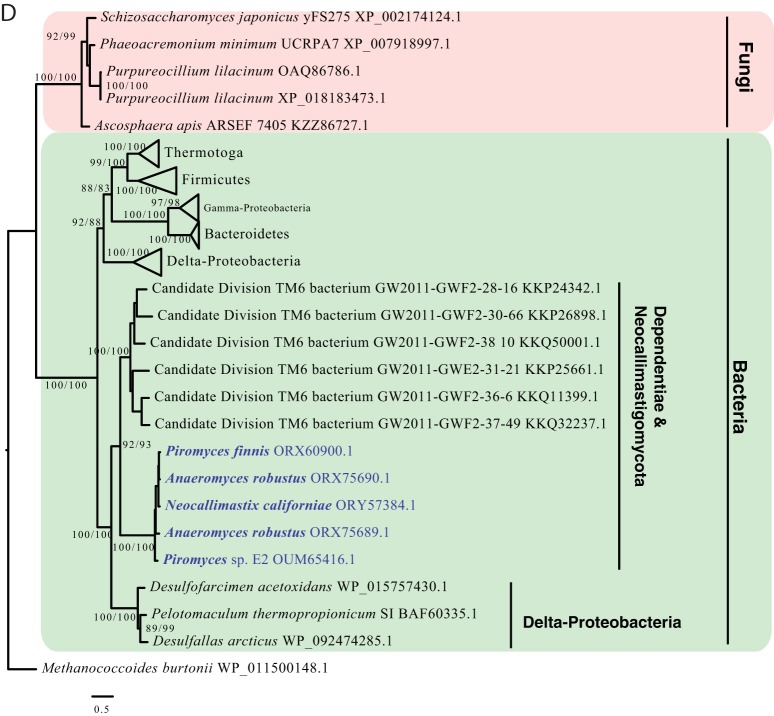
Similar articles
-
Molecular Dating of the Emergence of Anaerobic Rumen Fungi and the Impact of Laterally Acquired Genes.mSystems. 2019 Aug 27;4(4):e00247-19. doi: 10.1128/mSystems.00247-19. mSystems. 2019. PMID: 31455637 Free PMC article.
-
Seven new Neocallimastigomycota genera from wild, zoo-housed, and domesticated herbivores greatly expand the taxonomic diversity of the phylum.Mycologia. 2020 Nov-Dec;112(6):1212-1239. doi: 10.1080/00275514.2019.1696619. Epub 2020 Feb 14. Mycologia. 2020. PMID: 32057282
-
Anaerobic fungi in the tortoise alimentary tract illuminate early stages of host-fungal symbiosis and Neocallimastigomycota evolution.Nat Commun. 2024 Mar 28;15(1):2714. doi: 10.1038/s41467-024-47047-4. Nat Commun. 2024. PMID: 38548766 Free PMC article.
-
Anaerobic fungi (phylum Neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential.FEMS Microbiol Ecol. 2014 Oct;90(1):1-17. doi: 10.1111/1574-6941.12383. Epub 2014 Aug 11. FEMS Microbiol Ecol. 2014. PMID: 25046344 Review.
-
Horizontal gene transfer in fungi.FEMS Microbiol Lett. 2012 Apr;329(1):1-8. doi: 10.1111/j.1574-6968.2011.02465.x. Epub 2011 Dec 15. FEMS Microbiol Lett. 2012. PMID: 22112233 Review.
Cited by
-
Rumen Protozoa Play a Significant Role in Fungal Predation and Plant Carbohydrate Breakdown.Front Microbiol. 2020 Apr 29;11:720. doi: 10.3389/fmicb.2020.00720. eCollection 2020. Front Microbiol. 2020. PMID: 32411103 Free PMC article.
-
The anaerobic gut fungal community in ostriches (Struthio camelus).bioRxiv [Preprint]. 2025 Mar 30:2025.03.28.646006. doi: 10.1101/2025.03.28.646006. bioRxiv. 2025. PMID: 40196465 Free PMC article. Preprint.
-
Phylogenetic Analyses of Glycosyl Hydrolase Family 6 Genes in Tunicates: Possible Horizontal Transfer.Genes (Basel). 2020 Aug 13;11(8):937. doi: 10.3390/genes11080937. Genes (Basel). 2020. PMID: 32823766 Free PMC article.
-
A novel D-xylose isomerase from the gut of the wood feeding beetle Odontotaenius disjunctus efficiently expressed in Saccharomyces cerevisiae.Sci Rep. 2021 Feb 26;11(1):4766. doi: 10.1038/s41598-021-83937-z. Sci Rep. 2021. PMID: 33637780 Free PMC article.
-
HGT in the human and skin commensal Malassezia: A bacterially derived flavohemoglobin is required for NO resistance and host interaction.Proc Natl Acad Sci U S A. 2020 Jul 7;117(27):15884-15894. doi: 10.1073/pnas.2003473117. Epub 2020 Jun 23. Proc Natl Acad Sci U S A. 2020. PMID: 32576698 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
