

Predicting disease-causing variant combinations
- PMID: 31127050
- PMCID: PMC6575632
- DOI: 10.1073/pnas.1815601116
Predicting disease-causing variant combinations
Abstract
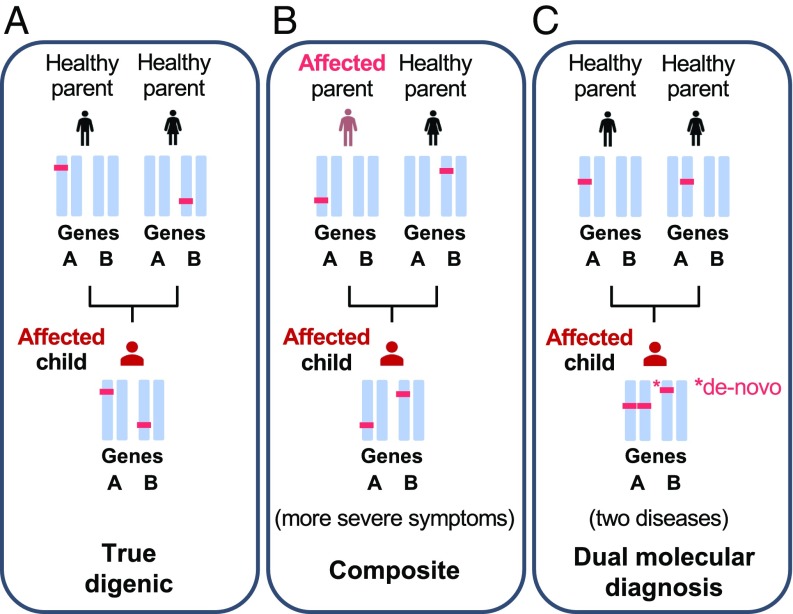
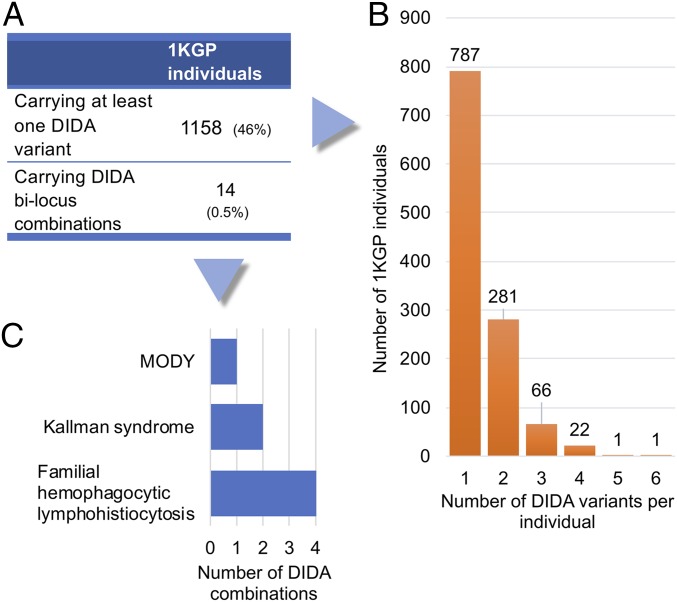
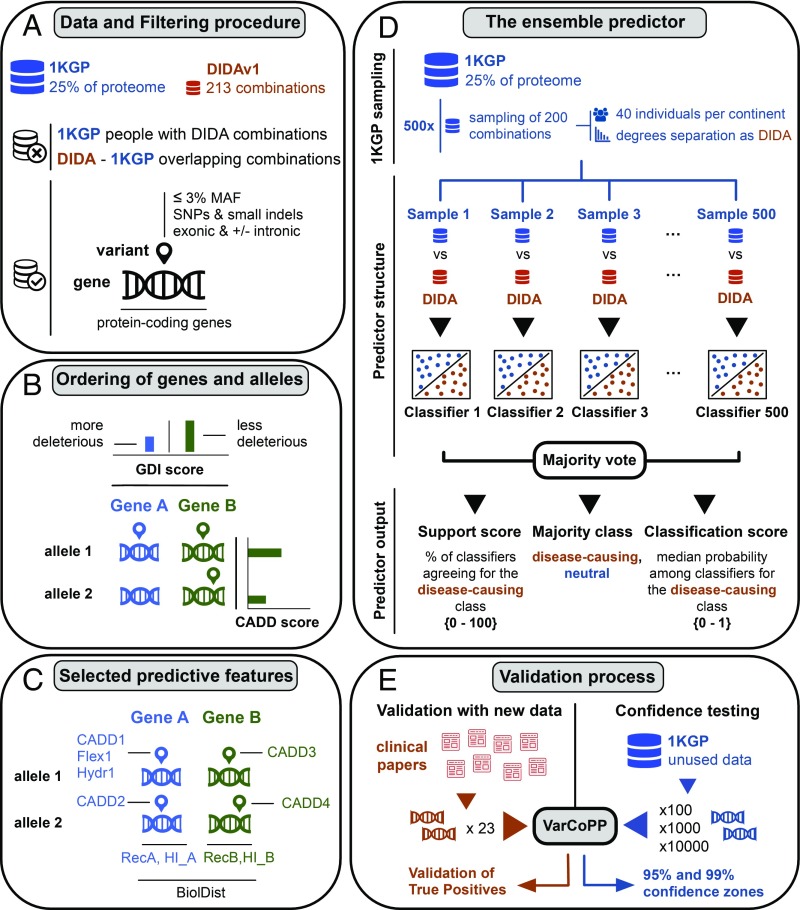
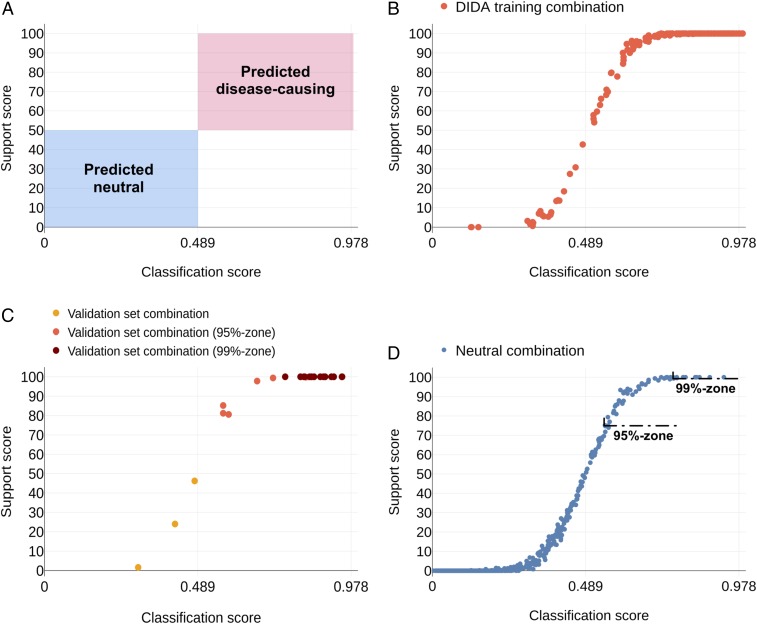
Notwithstanding important advances in the context of single-variant pathogenicity identification, novel breakthroughs in discerning the origins of many rare diseases require methods able to identify more complex genetic models. We present here the Variant Combinations Pathogenicity Predictor (VarCoPP), a machine-learning approach that identifies pathogenic variant combinations in gene pairs (called digenic or bilocus variant combinations). We show that the results produced by this method are highly accurate and precise, an efficacy that is endorsed when validating the method on recently published independent disease-causing data. Confidence labels of 95% and 99% are identified, representing the probability of a bilocus combination being a true pathogenic result, providing geneticists with rational markers to evaluate the most relevant pathogenic combinations and limit the search space and time. Finally, the VarCoPP has been designed to act as an interpretable method that can provide explanations on why a bilocus combination is predicted as pathogenic and which biological information is important for that prediction. This work provides an important step toward the genetic understanding of rare diseases, paving the way to clinical knowledge and improved patient care.
Keywords: bilocus combination; oligogenic; pathogenicity; prediction; variants.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
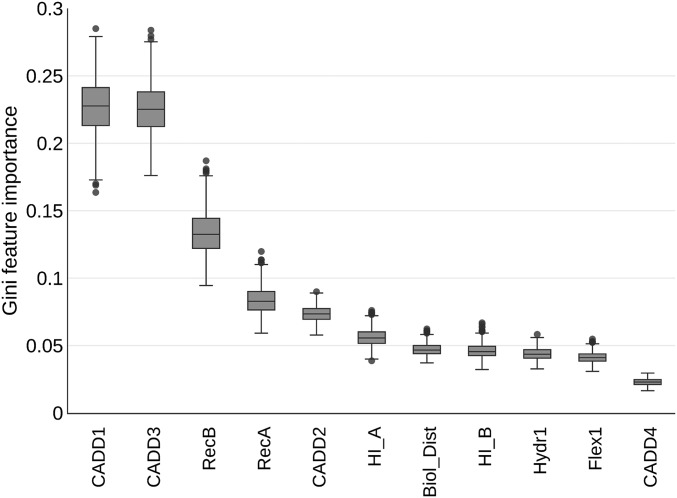
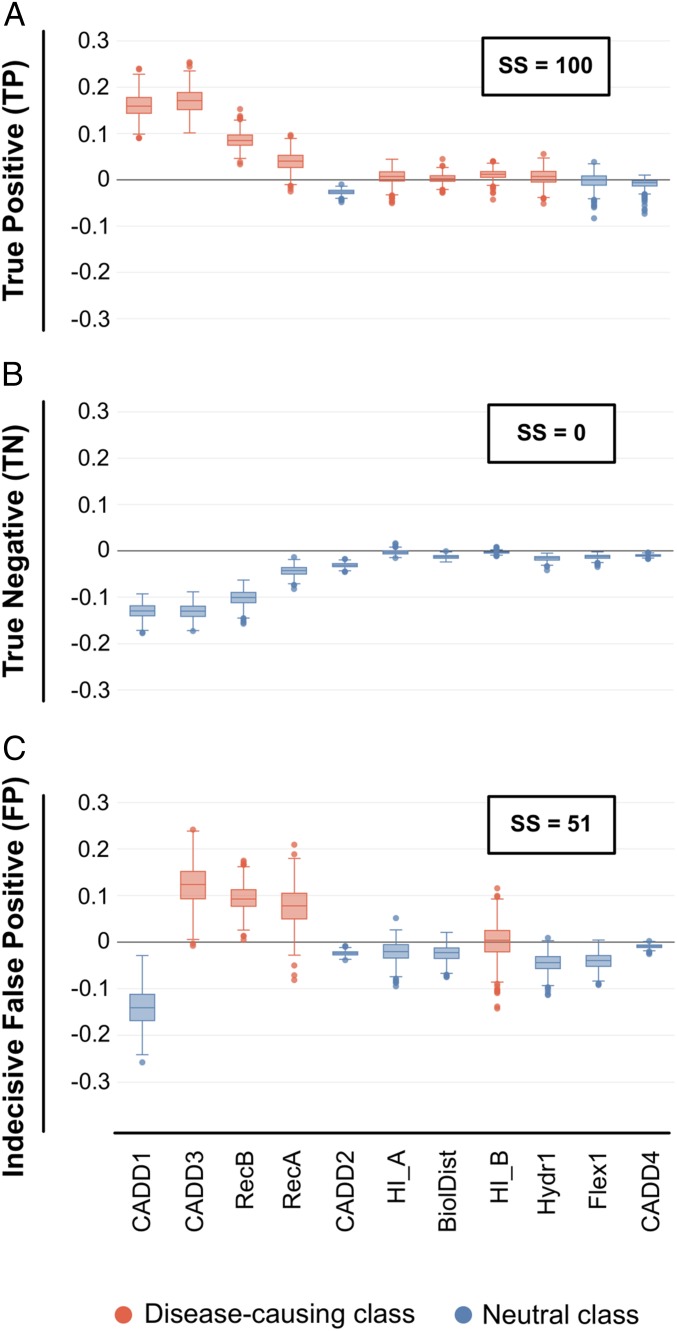
Figures
References
-
- NHLBI GO Exome Sequencing Project (ESP) , Exome Variant Server. http://evs.gs.washington.edu/EVS/. Accessed 15 May 2019.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
