

Microhomologies are prevalent at Cas9-induced larger deletions
- PMID: 31127293
- PMCID: PMC6698657
- DOI: 10.1093/nar/gkz459
Microhomologies are prevalent at Cas9-induced larger deletions
Abstract
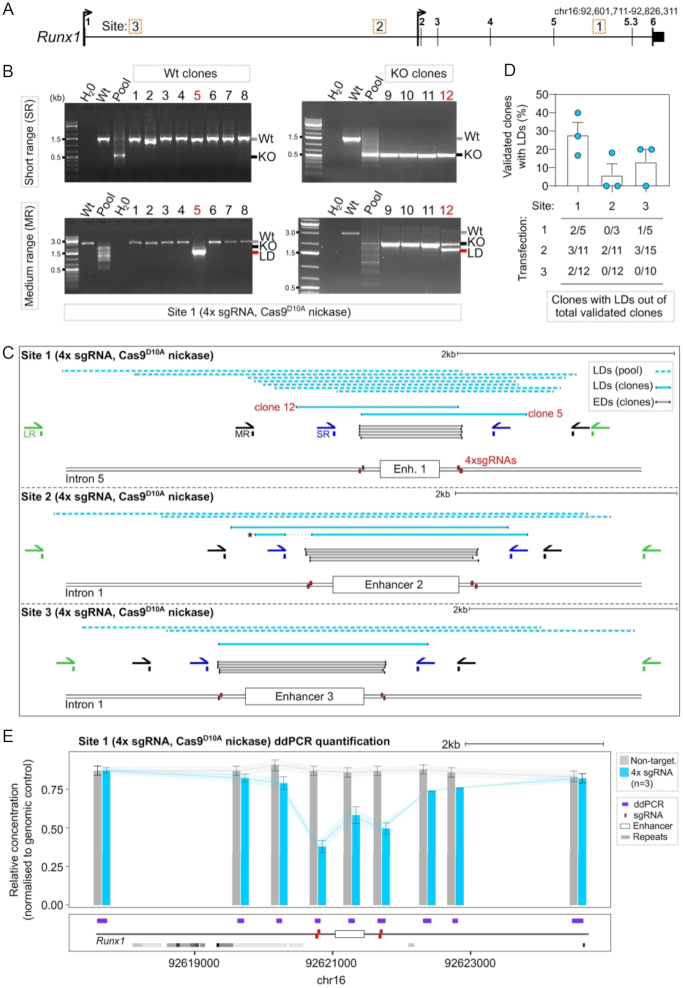
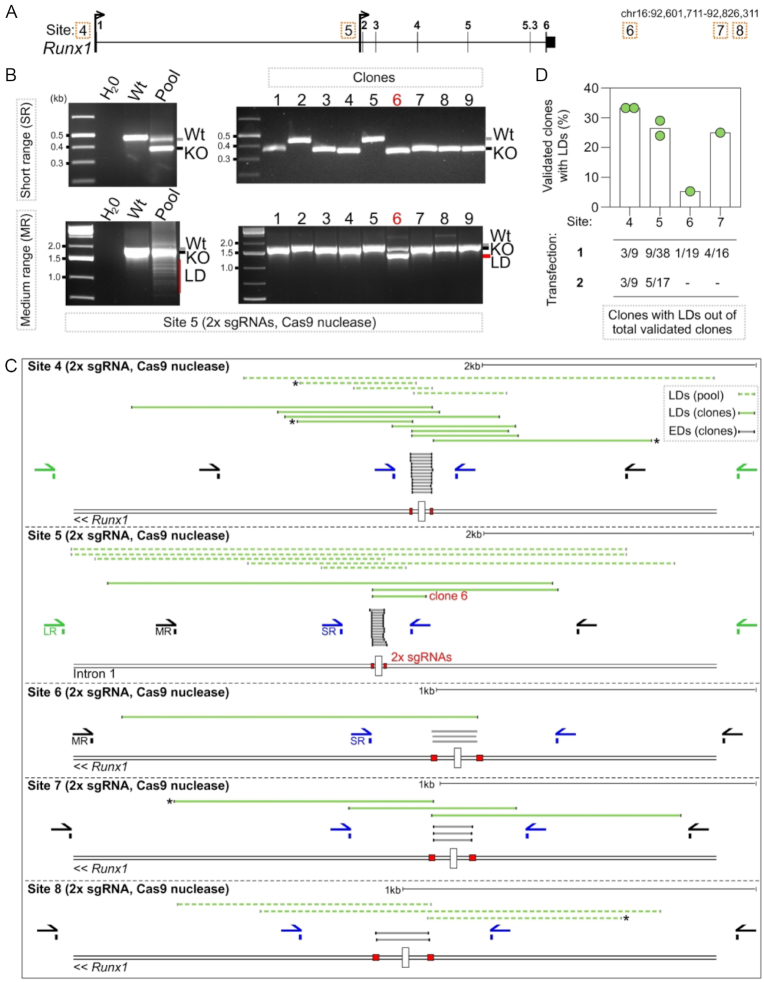
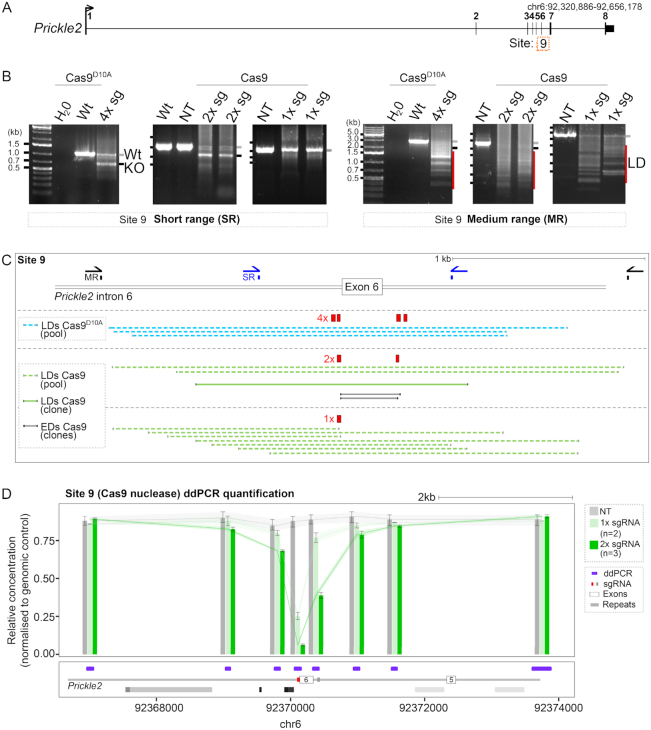
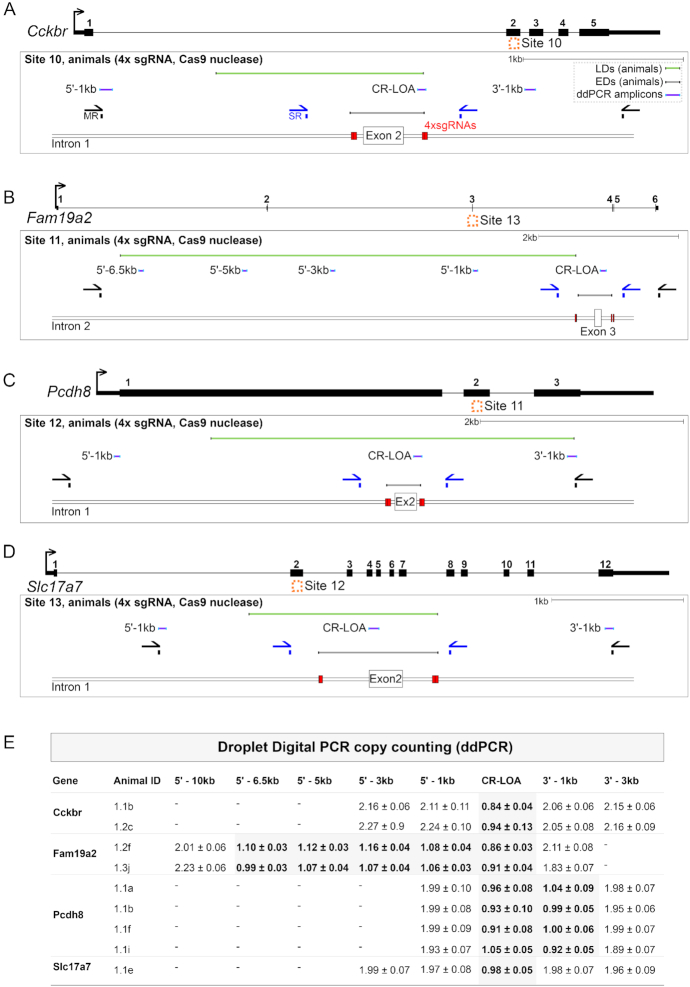
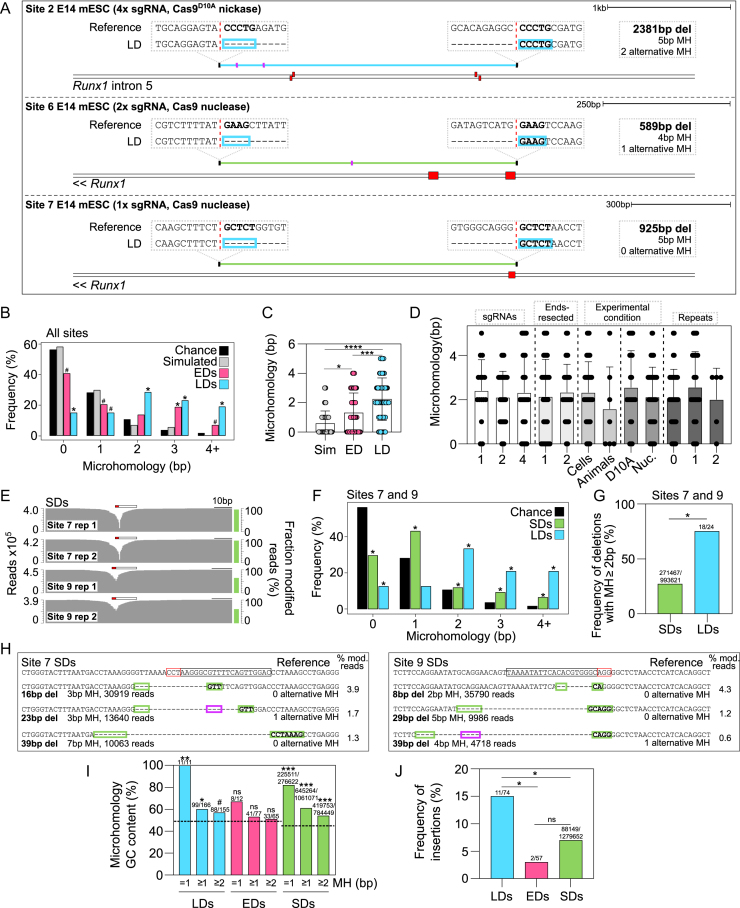
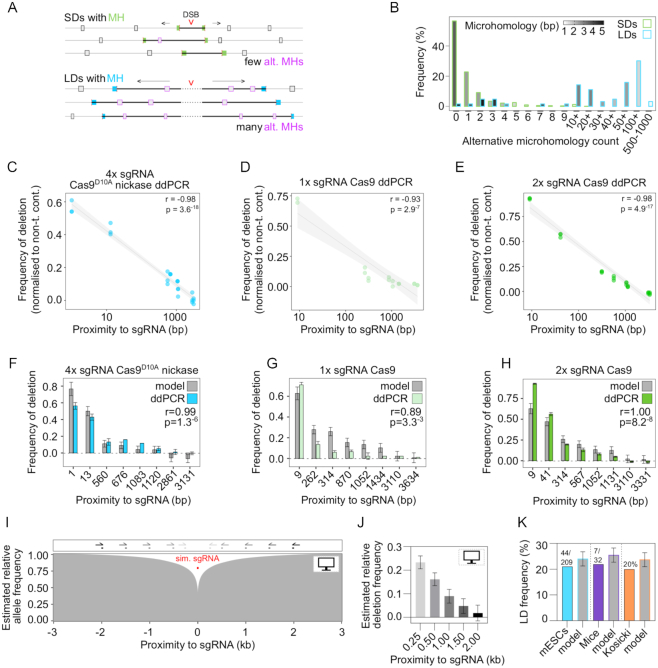
The CRISPR system is widely used in genome editing for biomedical research. Here, using either dual paired Cas9D10A nickases or paired Cas9 nuclease we characterize unintended larger deletions at on-target sites that frequently evade common genotyping practices. We found that unintended larger deletions are prevalent at multiple distinct loci on different chromosomes, in cultured cells and mouse embryos alike. We observed a high frequency of microhomologies at larger deletion breakpoint junctions, suggesting the involvement of microhomology-mediated end joining in their generation. In populations of edited cells, the distribution of larger deletion sizes is dependent on proximity to sgRNAs and cannot be predicted by microhomology sequences alone.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Caldecott K.W. DNA single-strand break repair. Exp. Cell Res. 2014; 329:2–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
