

A Bird's-Eye View of the Multiple Biochemical Mechanisms that Propel Pathology of Alzheimer's Disease: Recent Advances and Mechanistic Perspectives on How to Halt the Disease Progression Targeting Multiple Pathways
- PMID: 31127770
- PMCID: PMC6598003
- DOI: 10.3233/JAD-181230
A Bird's-Eye View of the Multiple Biochemical Mechanisms that Propel Pathology of Alzheimer's Disease: Recent Advances and Mechanistic Perspectives on How to Halt the Disease Progression Targeting Multiple Pathways
Abstract
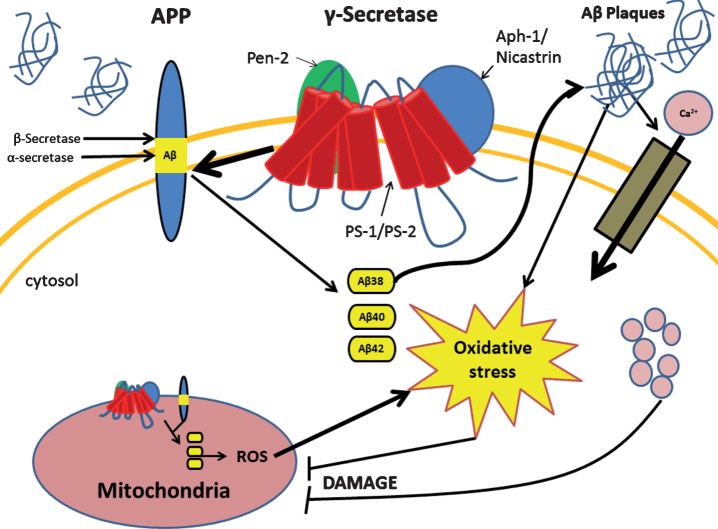
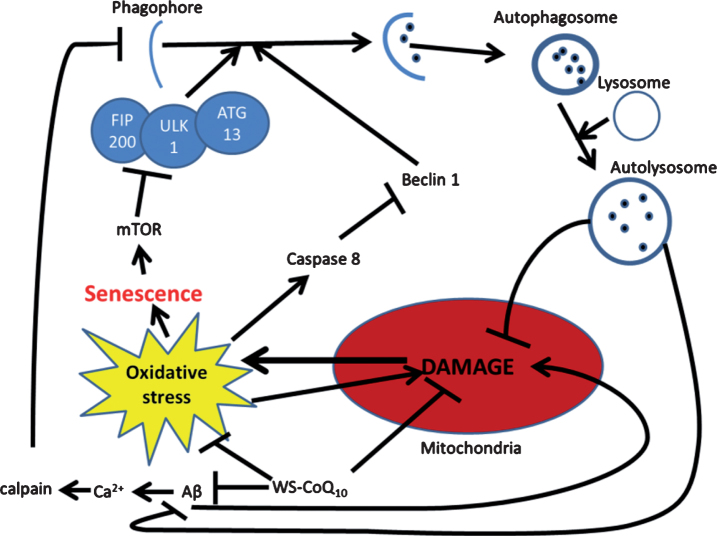
Neurons consume the highest amount of oxygen, depend on oxidative metabolism for energy, and survive for the lifetime of an individual. Therefore, neurons are vulnerable to death caused by oxidative-stress, accumulation of damaged and dysfunctional proteins and organelles. There is an exponential increase in the number of patients diagnosed with neurodegenerative diseases such as Alzheimer's (AD) as the number of elderly increases exponentially. Development of AD pathology is a complex phenomenon characterized by neuronal death, accumulation of extracellular amyloid-β plaques and neurofibrillary tangles, and most importantly loss of memory and cognition. These pathologies are most likely caused by mechanisms including oxidative stress, mitochondrial dysfunction/stress, accumulation of misfolded proteins, and defective organelles due to impaired proteasome and autophagy mechanisms. Currently, there are no effective treatments to halt the progression of this disease. In order to treat this complex disease with multiple biochemical pathways involved, a complex treatment regimen targeting different mechanisms should be investigated. Furthermore, as AD is a progressive disease-causing morbidity over many years, any chemo-modulator for treatment must be used over long period of time. Therefore, treatments must be safe and non-interfering with other processes. Ideally, a treatment like medicinal food or a supplement that can be taken regularly without any side effect capable of reducing oxidative stress, stabilizing mitochondria, activating autophagy or proteasome, and increasing energy levels of neurons would be the best solution. This review summarizes progress in research on different mechanisms of AD development and some of the potential therapeutic development strategies targeting the aforementioned pathologies.
Keywords: Alzheimer’s disease; amyloid-β plaques; ashwagandha; autophagy; mitochondrial dysfunction; oxidative stress; presenilin-1; water-soluble coenzyme-Q10.
Conflict of interest statement
Authors’ disclosures available online (
Figures
References
-
- Burns A, Iliffe S (2009) Alzheimer’s disease. BMJ 338, b158. - PubMed
-
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C (2009) Age, neuropathology, and dementia. N Engl J Med 360, 2302–2309. - PubMed
-
- Scheltens P, Blennow K, Breteler MMB, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM (2016) Alzheimer’s disease. Lancet 388, 505–517. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
