

Analysis of Spounaviruses as a Case Study for the Overdue Reclassification of Tailed Phages
- PMID: 31127947
- PMCID: PMC7409376
- DOI: 10.1093/sysbio/syz036
Analysis of Spounaviruses as a Case Study for the Overdue Reclassification of Tailed Phages
Abstract
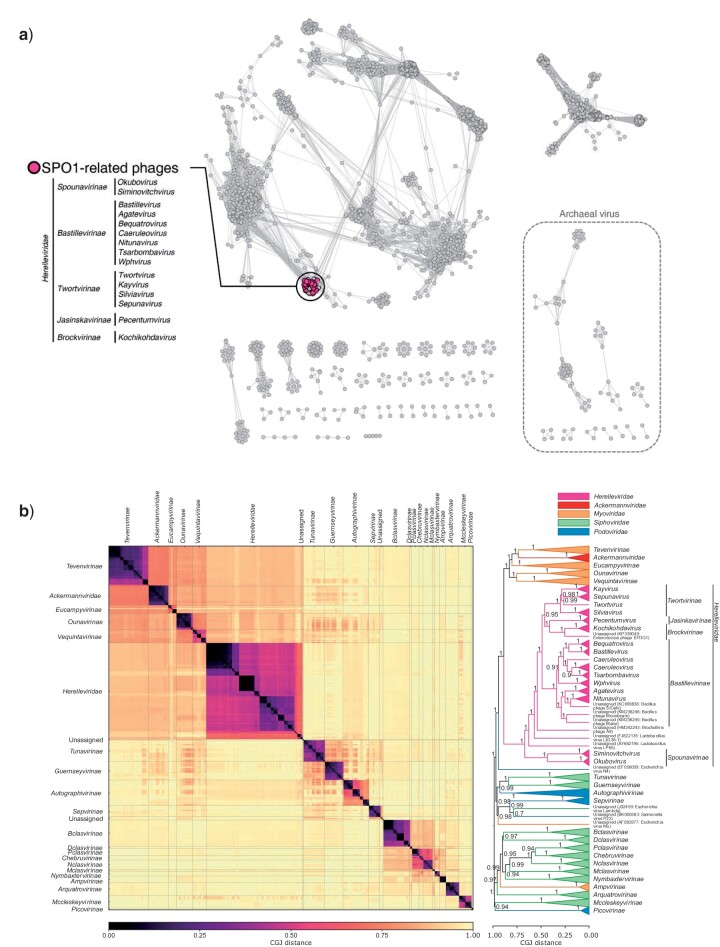
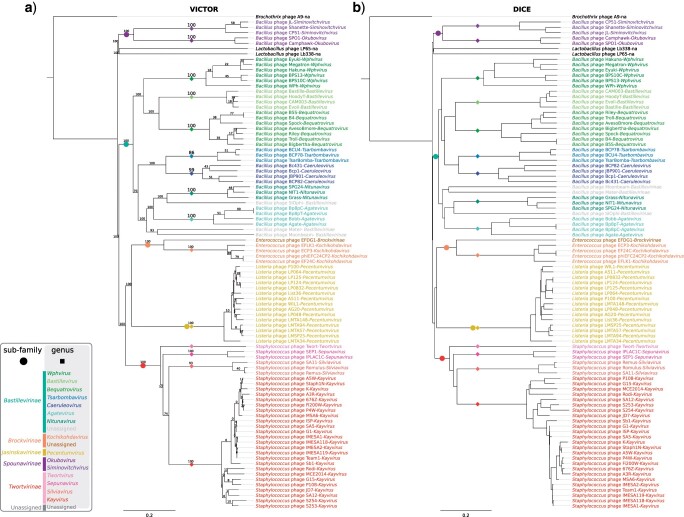
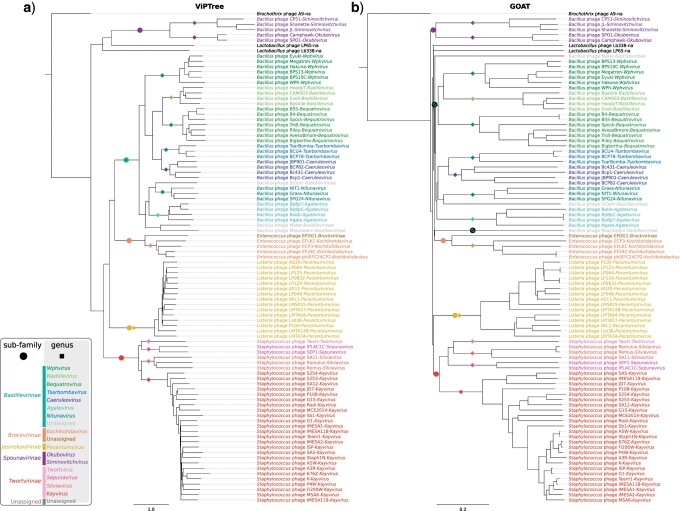
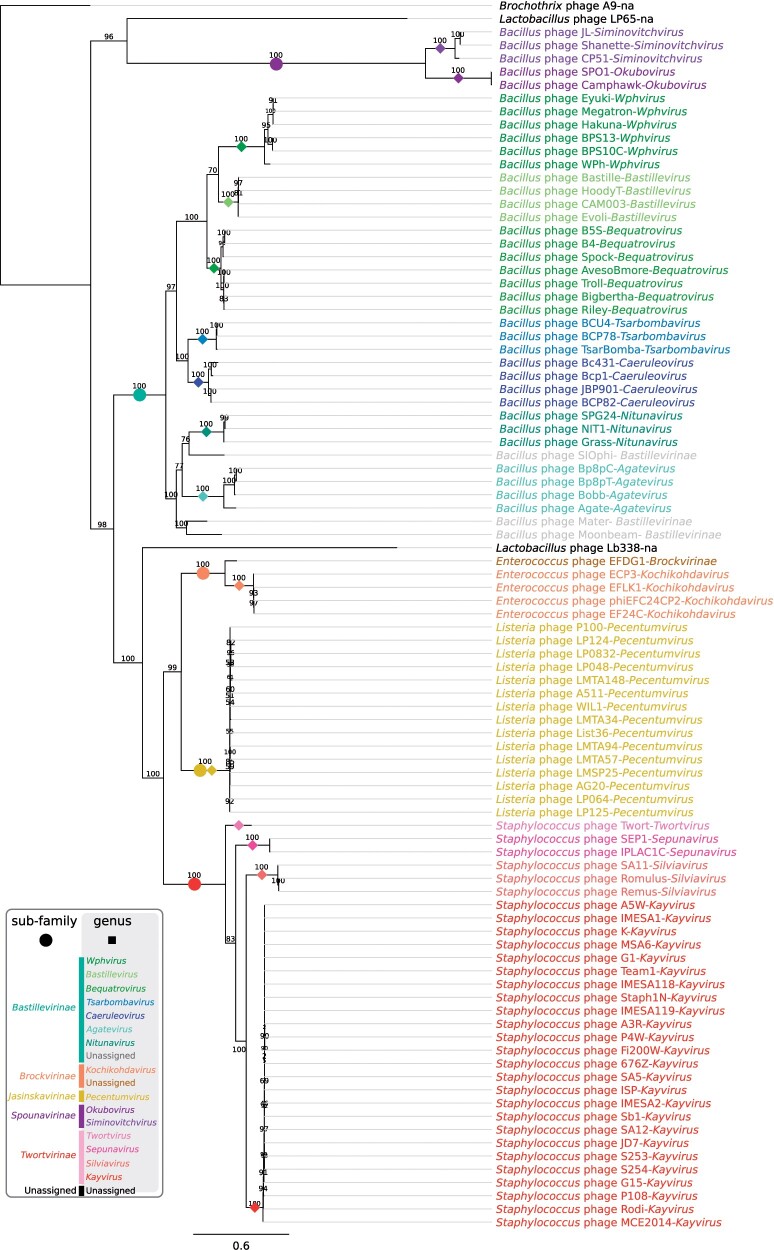
Tailed bacteriophages are the most abundant and diverse viruses in the world, with genome sizes ranging from 10 kbp to over 500 kbp. Yet, due to historical reasons, all this diversity is confined to a single virus order-Caudovirales, composed of just four families: Myoviridae, Siphoviridae, Podoviridae, and the newly created Ackermannviridae family. In recent years, this morphology-based classification scheme has started to crumble under the constant flood of phage sequences, revealing that tailed phages are even more genetically diverse than once thought. This prompted us, the Bacterial and Archaeal Viruses Subcommittee of the International Committee on Taxonomy of Viruses (ICTV), to consider overall reorganization of phage taxonomy. In this study, we used a wide range of complementary methods-including comparative genomics, core genome analysis, and marker gene phylogenetics-to show that the group of Bacillus phage SPO1-related viruses previously classified into the Spounavirinae subfamily, is clearly distinct from other members of the family Myoviridae and its diversity deserves the rank of an autonomous family. Thus, we removed this group from the Myoviridae family and created the family Herelleviridae-a new taxon of the same rank. In the process of the taxon evaluation, we explored the feasibility of different demarcation criteria and critically evaluated the usefulness of our methods for phage classification. The convergence of results, drawing a consistent and comprehensive picture of a new family with associated subfamilies, regardless of method, demonstrates that the tools applied here are particularly useful in phage taxonomy. We are convinced that creation of this novel family is a crucial milestone toward much-needed reclassification in the Caudovirales order.
Keywords: Caudovirales; Herelleviridae; phylogenetics; phylogenomics; spounavirus; virus classification; virus taxonomy.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society of Systematic Biologists.
Figures
References
-
- Adams M.J.,, Lefkowitz E.J.,, King A.M,, Harrach B.,, Harrison R.L.,, Knowles N.J.,, Kropinski A.M.,, Krupovic M.,, Kuhn J.H.,, Mushegian A.R.,, Nibert M.L.,, Sabanadzovic S.,, Sanfaçon H.,, Siddell S.G.,, Simmonds P.,, Varsani A.,, Zerbini F.M.,, Orton R.J.,, Smith D.B.,, Gorbalenya A.E.,, Davison A.J. 2017. 50 years of the International Committee on Taxonomy of Viruses: progress and prospects. Arch. Virol. 162:1441–1446. doi: 10.1007/s00705-016-3215-y. - DOI - PubMed
-
- Adriaenssens E.M.,, Ackermann H.-W.,, Anany H.,, Blasdel B.,, Connerton I.F.,, Goulding D.,, Griffiths M.W.,, Hooton S.P.,, Kutter E.M.,, Kropinski A.M.,, Lee J.-H.,, Maes M.,, Pickard D.,, Ryu S.,, Sepehrizadeh Z.,, Shahrbabak S.S.,, Toribio A.L.,, Lavigne R. 2012. A suggested new bacteriophage genus: “Viunalikevirus”. Arch. Virol. 157:2035–2046. doi: 10.1007/s00705-012-1360-5. - DOI - PMC - PubMed
-
- Adriaenssens E.M.,, Krupovic M.,, Knezevic P.,, Ackermann H.W.,, Barylski J.,, Brister J.R.,, Clokie M.R.,, Duffy S.,, Dutilh B.E.,, Edwards R.A.,, Enault F.,, Jang H.B.,, Klumpp J.,, Kropinski A.M.,, Lavigne R.,, Poranen M.M.,, Prangishvili D.,, Rumnieks J.,, Sullivan M.B.,, Wittmann J.,, Oksanen H.M.,, Gillis A.,, Kuhn J.H. 2017. Taxonomy of prokaryotic viruses: 2016 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 162:1153–1157. doi: 10.1007/s00705-016-3173-4. - DOI - PubMed
-
- Adriaenssens E.M.,, Wittmann J.,, Kuhn J.H.,, Turner D.,, Sullivan M.B.,, Dutilh B.E.,, Jang H.B.,, van Zyl L.J.,, Klumpp J.,, Lobocka M.,, Moreno Switt A.I.,, Rumnieks J.,, Edwards R.A.,, Uchiyama J.,, Alfenas-Zerbini P.,, Petty N.K.,, Kropinski A.M.,, Barylski J.,, Gillis A.,, Clokie M.R.J.,, Prangishvili D.,, Lavigne R.,, Aziz R.K.,, Duffy S.,, Krupovic M.,, Poranen M.M.,, Knezevic P.,, Enault F.,, Tong Y.,, Oksanen H.M.,, Brister J.R. 2018. Taxonomy of prokaryotic viruses: 2017 update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 163:1125–1129. doi: 10.1007/s00705-018-3723-z. - DOI - PubMed
