

Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies
- PMID: 31128155
- PMCID: PMC6708787
- DOI: 10.1016/j.pharmthera.2019.05.009
Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies
Abstract
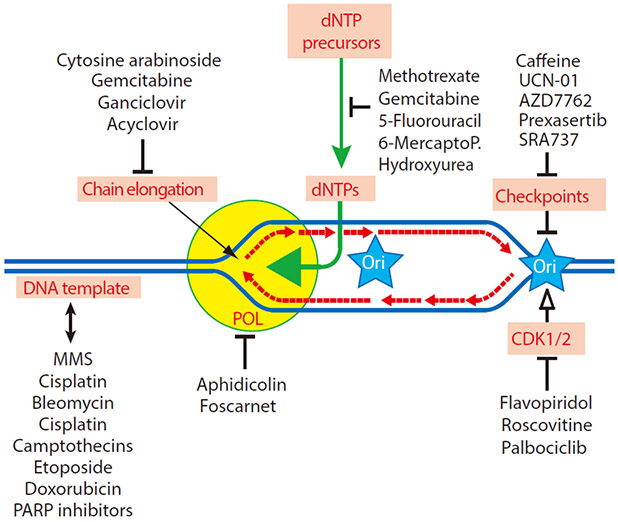
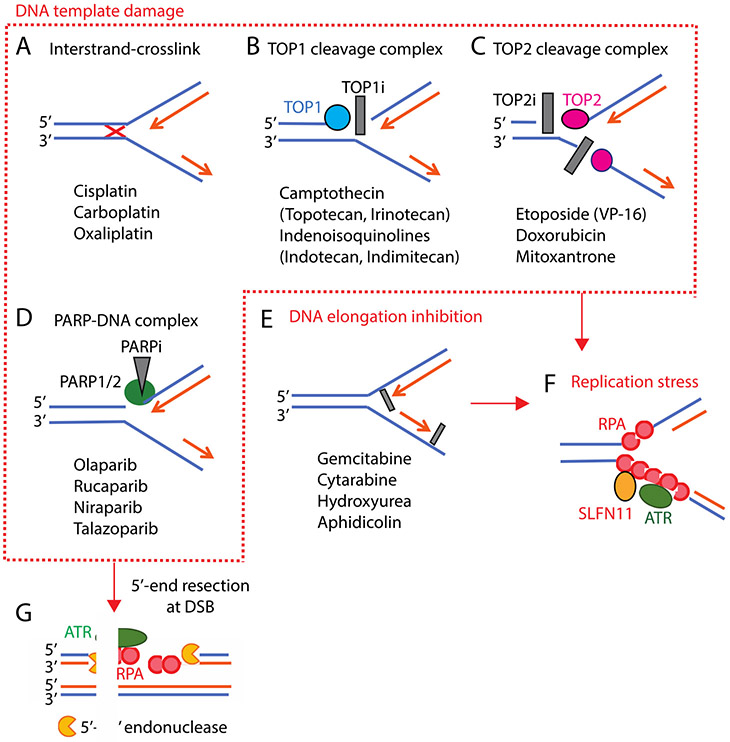
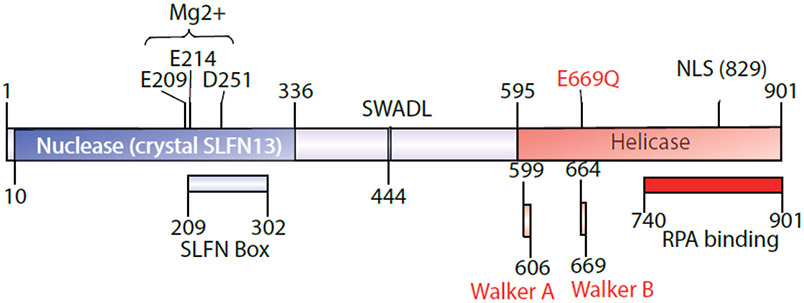
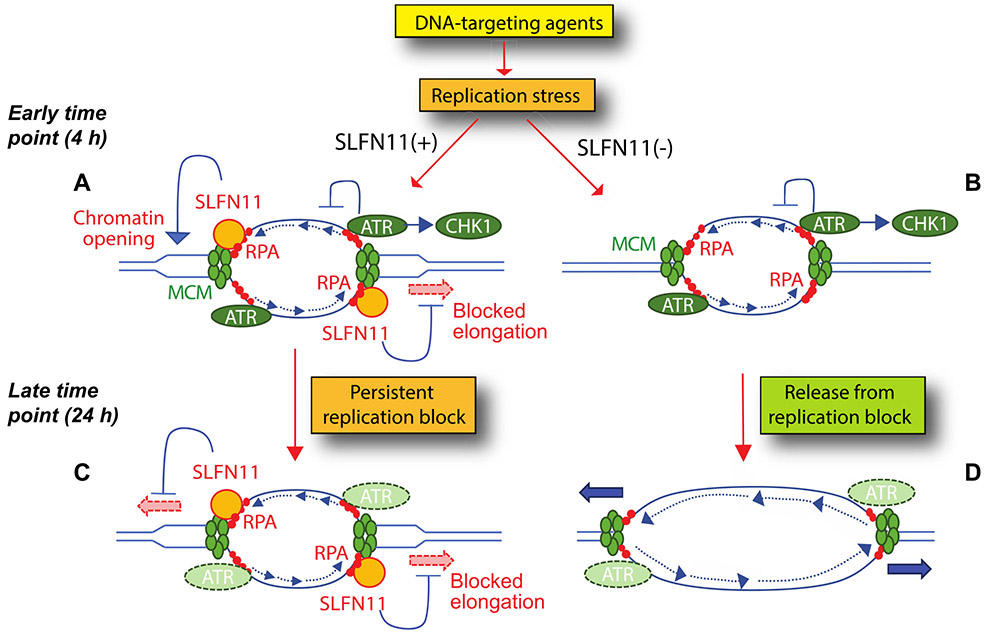
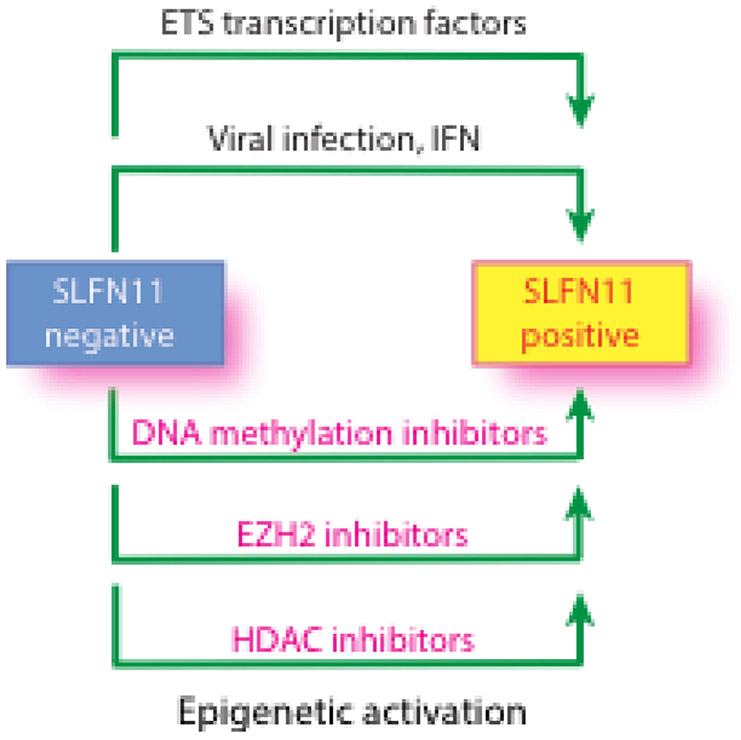
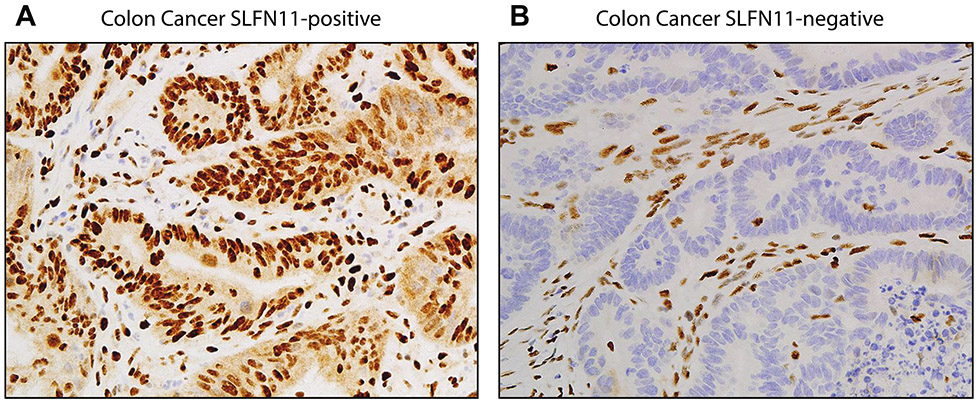
Schlafen 11 (SLFN11) sensitizes cells to a broad range of anti-cancer drugs including platinum derivatives (cisplatin and carboplatin), inhibitors of topoisomerases (irinotecan, topotecan, doxorubicin, daunorubicin, mitoxantrone and etoposide), DNA synthesis inhibitors (gemcitabine, cytarabine, hydroxyurea and nucleoside analogues), and poly(ADPribose) polymerase (PARP) inhibitors (olaparib, rucaparib, niraparib and talazoparib). In spite of their different primary mechanisms of action, all these drugs damage DNA during S-phase, activate the intra-S-phase checkpoint and induce replication fork slowing and stalling with single-stranded DNA segments coated with replication protein A. Such situation with abnormal replication forks is known as replication stress. SLFN11 irreversibly blocks replication in cells under replication stress, explaining why SLFN11-positive cells are markedly more efficiently killed by DNA-targeting drugs than SLFN11-negative cells. SLFN11 is inactivated in ~50% of cancer cell lines and in a large fraction of tumors, and is linked with the native immune, interferon and T-cells responses, implying the translational relevance of measuring SLFN11 expression as a predictive biomarker of response and resistance in patients. SLFN11 is also a plausible epigenetic target for reactivation by inhibitors of histone deacetylases (HDAC), DNA methyltransferases (DNMT) and EZH2 histone methyltransferase and for combination of these epigenetic inhibitors with DNA-targeting drugs in cells lacking SLFN11 expression. In addition, resistance due to lack of SLFN11 expression in tumors is a potential indication for cell-cycle checkpoint inhibitors in combination with DNA-targeting therapies.
Keywords: ATR; Cell cycle checkpoint; DNA damage response; DNA-targeting agent; Drug resistance; PARP inhibitors; Replication stress; SLFN11; Schlafen 11; Topoisomerases.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest
The authors have no conflicts of interest.
Figures
Similar articles
-
Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors.Clin Cancer Res. 2018 Apr 15;24(8):1944-1953. doi: 10.1158/1078-0432.CCR-17-0443. Epub 2018 Feb 1. Clin Cancer Res. 2018. PMID: 29391350 Free PMC article.
-
SLFN11 promotes CDT1 degradation by CUL4 in response to replicative DNA damage, while its absence leads to synthetic lethality with ATR/CHK1 inhibitors.Proc Natl Acad Sci U S A. 2021 Feb 9;118(6):e2015654118. doi: 10.1073/pnas.2015654118. Proc Natl Acad Sci U S A. 2021. PMID: 33536335 Free PMC article.
-
Immunohistochemical analysis of SLFN11 expression uncovers potential non-responders to DNA-damaging agents overlooked by tissue RNA-seq.Virchows Arch. 2021 Mar;478(3):569-579. doi: 10.1007/s00428-020-02840-6. Epub 2020 May 30. Virchows Arch. 2021. PMID: 32474729 Free PMC article.
-
Reconsidering the mechanisms of action of PARP inhibitors based on clinical outcomes.Cancer Sci. 2022 Sep;113(9):2943-2951. doi: 10.1111/cas.15477. Epub 2022 Jul 16. Cancer Sci. 2022. PMID: 35766436 Free PMC article. Review.
-
From predictive biomarker to therapeutic target: the dual role of SLFN11 in chemotherapy sensitivity.Cancer Chemother Pharmacol. 2025 Jun 18;95(1):60. doi: 10.1007/s00280-025-04781-w. Cancer Chemother Pharmacol. 2025. PMID: 40531330 Review.
Cited by
-
Involvement of Met receptor pathway in aggressive behavior of colorectal cancer cells induced by parathyroid hormone-related peptide.World J Gastroenterol. 2022 Jul 14;28(26):3177-3200. doi: 10.3748/wjg.v28.i26.3177. World J Gastroenterol. 2022. PMID: 36051345 Free PMC article.
-
Candidate biomarker assessment for pharmacological response.Transl Oncol. 2020 Oct;13(10):100830. doi: 10.1016/j.tranon.2020.100830. Epub 2020 Jul 8. Transl Oncol. 2020. PMID: 32652468 Free PMC article.
-
Schlafen 11 further sensitizes BRCA-deficient cells to PARP inhibitors through single-strand DNA gap accumulation behind replication forks.Oncogene. 2024 Aug;43(32):2475-2489. doi: 10.1038/s41388-024-03094-1. Epub 2024 Jul 3. Oncogene. 2024. PMID: 38961202 Free PMC article.
-
Schlafen family member 11 indicates favorable prognosis of patients with head and neck cancer following platinum-based chemoradiotherapy.Front Oncol. 2023 Jan 19;12:978875. doi: 10.3389/fonc.2022.978875. eCollection 2022. Front Oncol. 2023. PMID: 36741698 Free PMC article.
-
SLFN5-mediated chromatin dynamics sculpt higher-order DNA repair topology.Mol Cell. 2023 Apr 6;83(7):1043-1060.e10. doi: 10.1016/j.molcel.2023.02.004. Epub 2023 Feb 27. Mol Cell. 2023. PMID: 36854302 Free PMC article.
References
-
- Agelopoulos K, Richter GH, Schmidt E, Dirksen U, von Heyking K, Moser B, Klein HU, Kontny U, Dugas M, Poos K, Korsching E, Buch T, Weckesser M, Schulze I, Besoke R, Witten A, Stoll M, Kohler G, Hartmann W, Wardelmann E, Rossig C, Baumhoer D, Jurgens H, Burdach S, Berdel WE, & Muller-Tidow C (2015). Deep Sequencing in Conjunction with Expression and Functional Analyses Reveals Activation of FGFR1 in Ewing Sarcoma. Clin Cancer Res, 21, 4935–4946. - PubMed
-
- Antony S, Agama KK, Miao ZH, Takagi K, Wright MH, Robles AI, Varticovski L, Nagarajan M, Morrell A, Cushman M, & Pommier Y (2007). Novel indenoisoquinolines NSC 725776 and NSC 724998 produce persistent topoisomerase I cleavage complexes and overcome multidrug resistance. Cancer research, 67, 10397–10405. - PubMed
-
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, & Garraway LA (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. - PMC - PubMed
-
- Branzei D, & Foiani M (2008). Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol, 9, 297–308. - PubMed
-
- Branzei D, & Foiani M (2010). Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol, 11, 208–219. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
