

Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review
- PMID: 31130681
- PMCID: PMC6572054
- DOI: 10.3390/jcm8050750
Molecular Genetics and Interferon Signature in the Italian Aicardi Goutières Syndrome Cohort: Report of 12 New Cases and Literature Review
Abstract
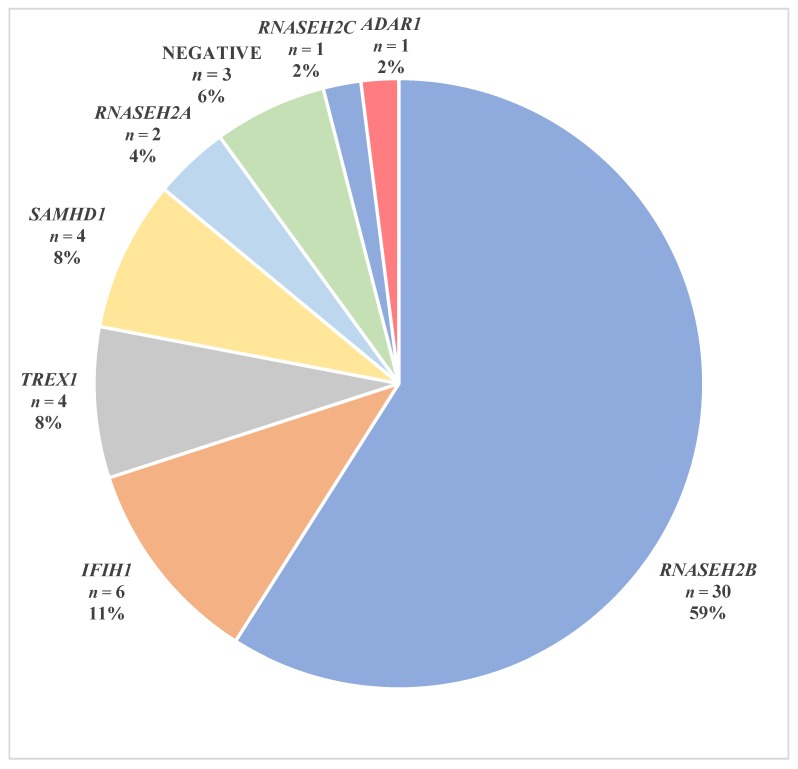
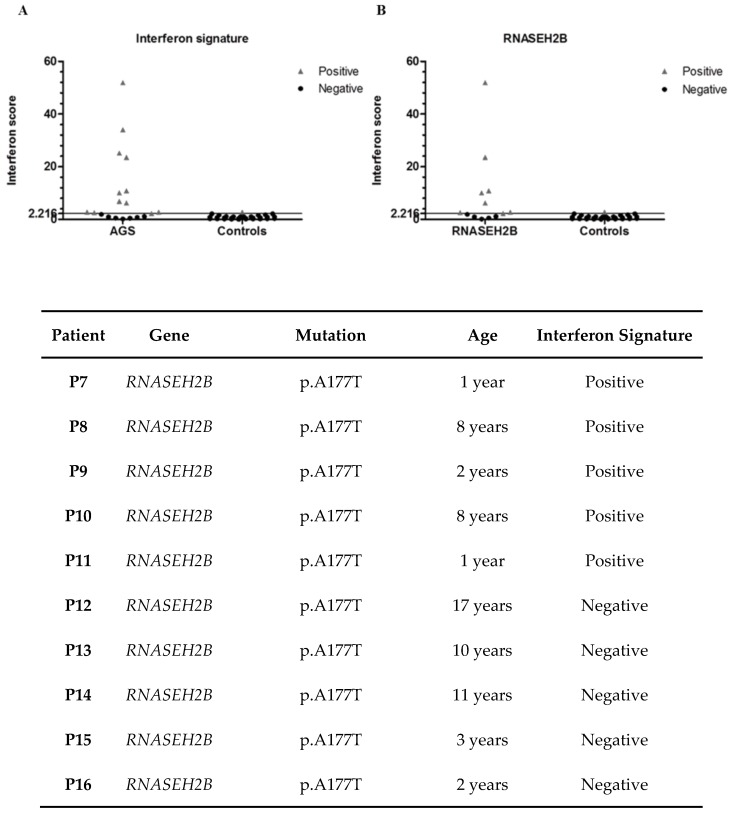
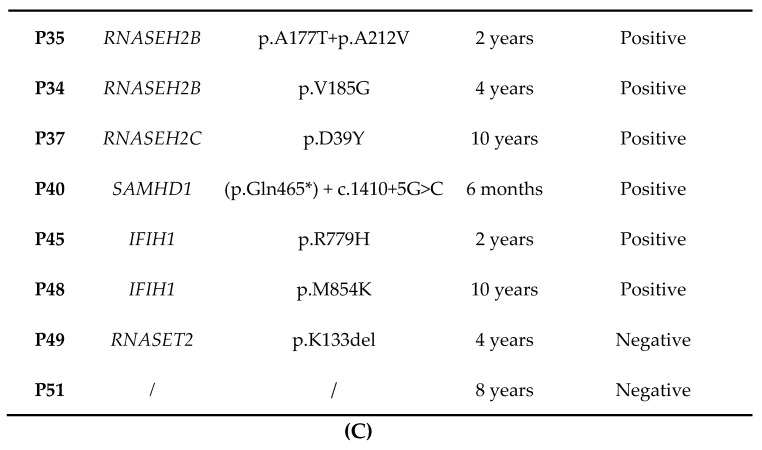
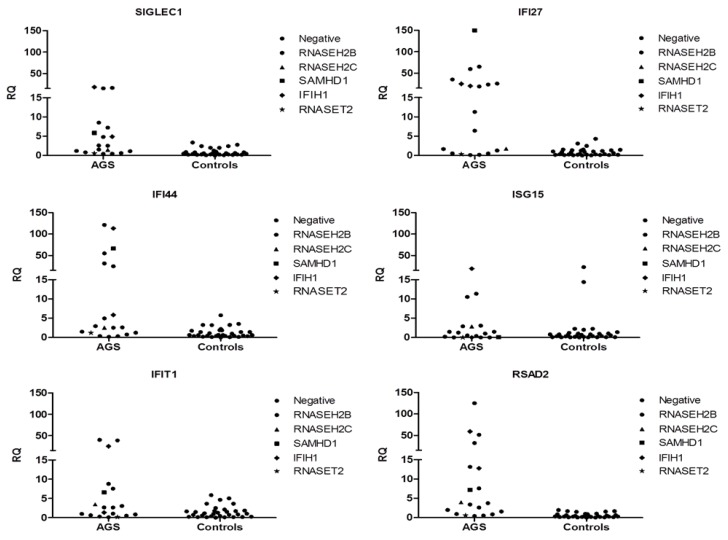
Aicardi-Goutières syndrome (AGS) is a genetically determined early onset encephalopathy characterized by cerebral calcification, leukodystrophy, and increased expression of interferon-stimulated genes (ISGs). Up to now, seven genes (TREX1, RNASEH2B, RNASEH2C, RNASEH2A, ADAR1, SAMHD1, IFIH1) have been associated with an AGS phenotype. Next Generation Sequencing (NGS) analysis was performed on 51 AGS patients and interferon signature (IS) was investigated in 18 AGS patients and 31 healthy controls. NGS identified mutations in 48 of 51 subjects, with three patients demonstrating a typical AGS phenotype but not carrying mutations in known AGS-related genes. Five mutations, in RNASEH2B, SAMHD1 and IFIH1 gene, were not previously reported. Eleven patients were positive and seven negatives for the upregulation of interferon signaling (IS > 2.216). This work presents, for the first time, the genetic data of an Italian cohort of AGS patients, with a higher percentage of mutations in RNASEH2B and a lower frequency of mutations in TREX1 than those seen in international series. RNASEH2B mutated patients showed a prevalence of negative IS consistent with data reported in the literature. We also identified five novel pathogenic mutations that warrant further functional investigation. Exome/genome sequencing will be performed in future studies in patients without a mutation in AGS-related genes.
Keywords: Aicardi-Goutières Syndrome; Interferon signature; Next Generation Sequencing.
Conflict of interest statement
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Figures
References
-
- Crow Y.J., Chase D.S., Lowenstein Schmidt J., Szynkiewicz M., Forte G.M., Gornall H.L., Oojageer A., Anderson B., Pizzino A., Helman G., et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A. 2015;167:296–312. doi: 10.1002/ajmg.a.36887. - DOI - PMC - PubMed
-
- Vanderver A., Prust M., Kadom N., Demarest S., Crow Y.J., Helman G., Orcesi S., La Piana R., Uggetti C., Wang J., et al. Early-Onset Aicardi-Goutières Syndrome: Magnetic Resonance Imaging (MRI) Pattern Recognition. J. Child. Neurol. 2015;30:1343–1348. doi: 10.1177/0883073814562252. - DOI - PMC - PubMed
-
- Fazzi E., Cattalini M., Orcesi S., Tincani A., Andreoli L., Balottin U., De Simone M., Fredi M., Facchetti F., Galli J., et al. Aicardi-Goutieres syndrome, a rare neurological disease in children: A new autoimmune disorder? Autoimmun. Rev. 2013;12:506–509. doi: 10.1016/j.autrev.2012.08.012. - DOI - PubMed
-
- Livingston J.H., Crow Y.J. Neurologic Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières Syndrome and Beyond. Neuropediatrics. 2016;47:355–360. - PubMed
