

Dysfunction of Cellular Proteostasis in Parkinson's Disease
- PMID: 31133790
- PMCID: PMC6524622
- DOI: 10.3389/fnins.2019.00457
Dysfunction of Cellular Proteostasis in Parkinson's Disease
Abstract
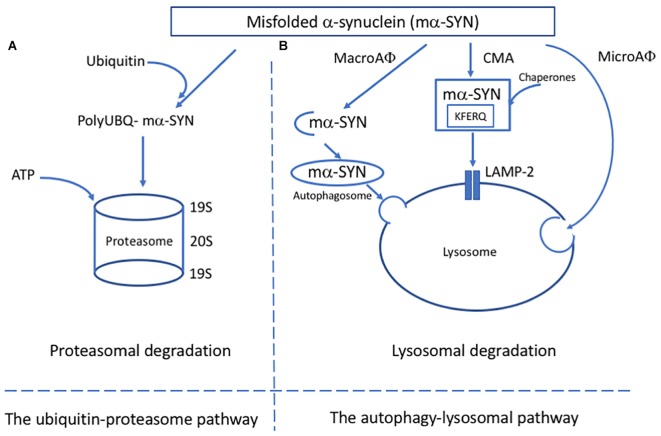
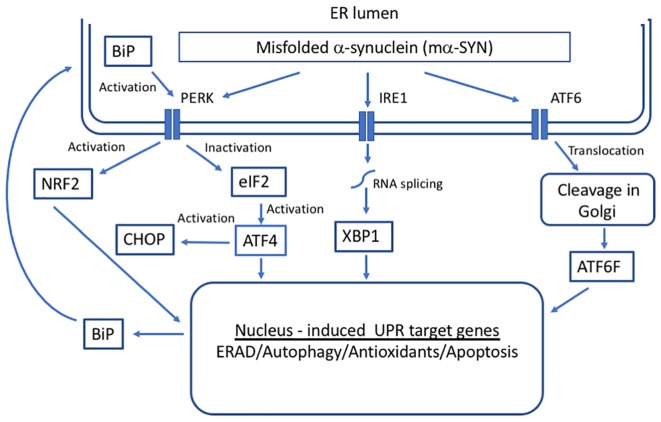
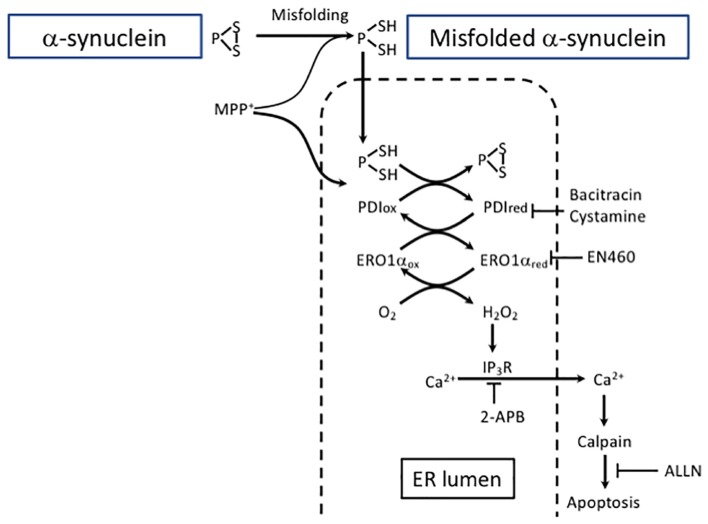
Despite decades of research, current therapeutic interventions for Parkinson's disease (PD) are insufficient as they fail to modify disease progression by ameliorating the underlying pathology. Cellular proteostasis (protein homeostasis) is an essential factor in maintaining a persistent environment for neuronal activity. Proteostasis is ensured by mechanisms including regulation of protein translation, chaperone-assisted protein folding and protein degradation pathways. It is generally accepted that deficits in proteostasis are linked to various neurodegenerative diseases including PD. While the proteasome fails to degrade large protein aggregates, particularly alpha-synuclein (α-SYN) in PD, drug-induced activation of autophagy can efficiently remove aggregates and prevent degeneration of dopaminergic (DA) neurons. Therefore, maintenance of these mechanisms is essential to preserve all cellular functions relying on a correctly folded proteome. The correlations between endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) that aims to restore proteostasis within the secretory pathway are well-established. However, while mild insults increase the activity of chaperones, prolonged cell stress, or insufficient adaptive response causes cell death. Modulating the activity of molecular chaperones, such as protein disulfide isomerase which assists refolding and contributes to the removal of unfolded proteins, and their associated pathways may offer a new approach for disease-modifying treatment. Here, we summarize some of the key concepts and emerging ideas on the relation of protein aggregation and imbalanced proteostasis with an emphasis on PD as our area of main expertise. Furthermore, we discuss recent insights into the strategies for reducing the toxic effects of protein unfolding in PD by targeting the ER UPR pathway.
Keywords: ER stress; UPR; alpha-synuclein; protein disulfide isomerase; proteostasis; refolding.
Figures
References
-
- Ambrosi G., Ghezzi C., Zangaglia R., Levandis G., Pacchetti C., Blandini F. (2015). Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 82 235–242. 10.1016/j.nbd.2015.06.008 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous