

Genome Analysis of Coxsackievirus A4 Isolates From Hand, Foot, and Mouth Disease Cases in Shandong, China
- PMID: 31134033
- PMCID: PMC6513881
- DOI: 10.3389/fmicb.2019.01001
Genome Analysis of Coxsackievirus A4 Isolates From Hand, Foot, and Mouth Disease Cases in Shandong, China
Abstract
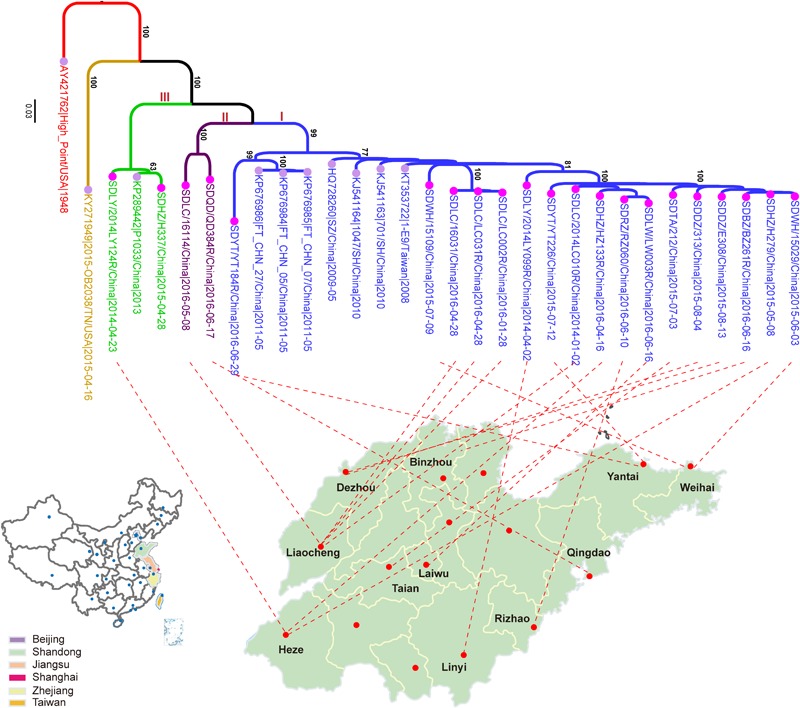
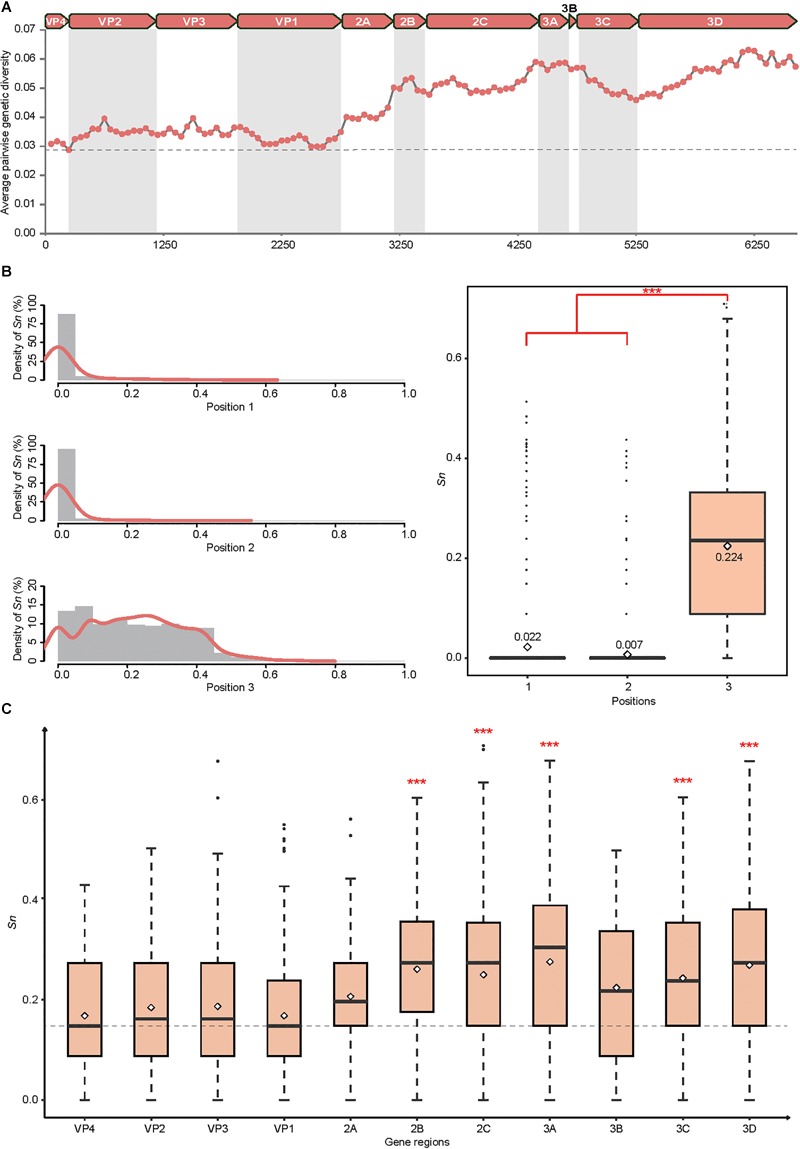
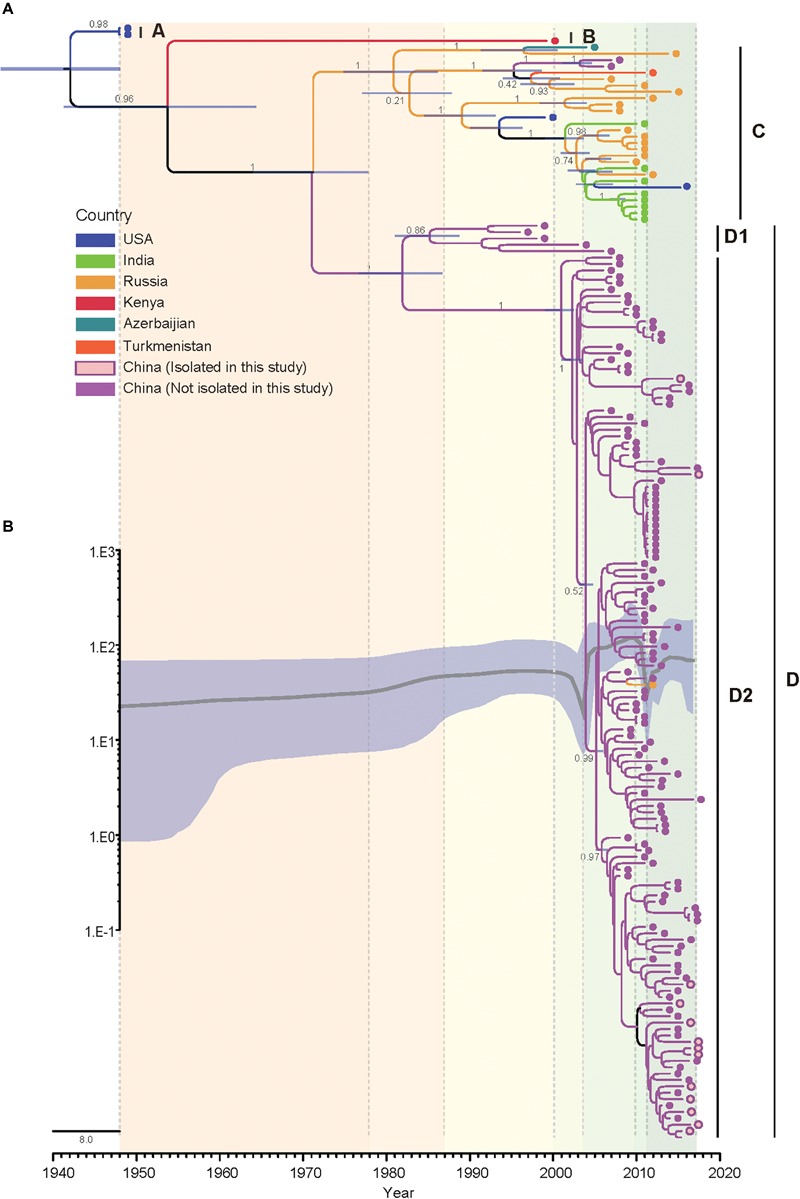
Coxsackievirus A4 (CVA4) is one of the most prevalent pathogens associated with hand, foot and mouth disease (HFMD), an acute febrile illness in children, and is also associated with acute localized exanthema, myocarditis, hepatitis and pancreatitis. Despite this, limited CVA4 genome sequences are currently available. Herein, complete genome sequences from CVA4 strains (n = 21), isolated from patients with HFMD in Shandong province, China between 2014 and 2016, were determined and phylogenetically characterized. Phylogenetic analysis of the VP1 gene from a larger CVA4 collection (n = 175) showed that CVA4 has evolved into four separable genotypes: A, B, C, and D; and genotype D could be further classified in to two sub-genotypes: D1 and D2. Each of the 21 newly described genomes derived from isolates that segregated with sub-genotype D2. The CVA4 genomes displayed significant intra-genotypic genetic diversity with frequent synonymous substitutions occurring at the third codon positions, particularly within the P2 region. However, VP1 was relatively stable and therefore represents a potential target for molecular diagnostics assays and also for the rational design of vaccine epitopes. The substitution rate of VP1 was estimated to be 5.12 × 10-3 substitutions/site/year, indicative of ongoing CVA4 evolution. Mutations at amino acid residue 169 in VP1 gene may be responsible for differing virulence of CVA4 strains. Bayesian skyline plot analysis showed that the population size of CVA4 has experienced several dynamic fluctuations since 1948. In summary, we describe the phylogenetic and molecular characterization of 21 complete genomes from CVA4 isolates which greatly enriches the known genomic diversity of CVA4 and underscores the need for further surveillance of CVA4 in China.
Keywords: Coxsackievirus A4; VP1; and mouth disease; foot; genotypes; hand; phylogenetic analysis.
Figures
References
-
- Cai C., Yao X., Zhuo F., He Y., Yang G. (2015). [Gene characterization of the VP1 region of Coxsackievirus A4 from herpangina cases in Shenzhen of China]. Zhonghua Yi Xue Za Zhi 95 1226–1229. - PubMed
-
- Carey D. E., Myers R. M. (1969). Isolation of type A4 coxsackie virus from the blood serum of a child with rapidly fatal illness marked by severe central nervous system manifestations. Indian J. Med. Res. 57 765–769. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources