

Long-Read Sequencing Emerging in Medical Genetics
- PMID: 31134132
- PMCID: PMC6514244
- DOI: 10.3389/fgene.2019.00426
Long-Read Sequencing Emerging in Medical Genetics
Abstract
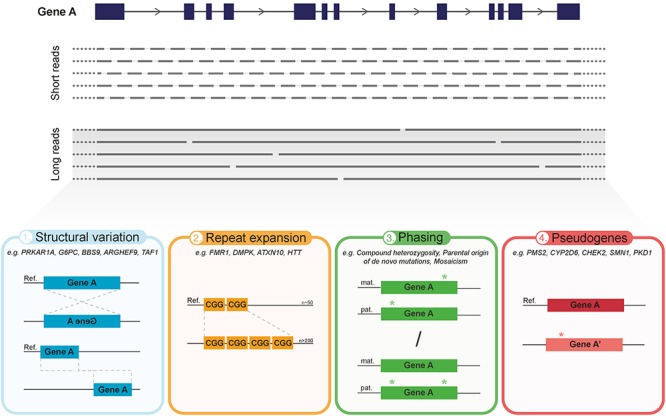
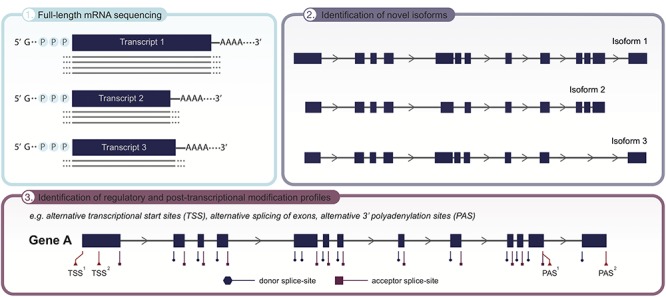
The wide implementation of next-generation sequencing (NGS) technologies has revolutionized the field of medical genetics. However, the short read lengths of currently used sequencing approaches pose a limitation for the identification of structural variants, sequencing repetitive regions, phasing of alleles and distinguishing highly homologous genomic regions. These limitations may significantly contribute to the diagnostic gap in patients with genetic disorders who have undergone standard NGS, like whole exome or even genome sequencing. Now, the emerging long-read sequencing (LRS) technologies may offer improvements in the characterization of genetic variation and regions that are difficult to assess with the prevailing NGS approaches. LRS has so far mainly been used to investigate genetic disorders with previously known or strongly suspected disease loci. While these targeted approaches already show the potential of LRS, it remains to be seen whether LRS technologies can soon enable true whole genome sequencing routinely. Ultimately, this could allow the de novo assembly of individual whole genomes used as a generic test for genetic disorders. In this article, we summarize the current LRS-based research on human genetic disorders and discuss the potential of these technologies to facilitate the next major advancements in medical genetics.
Keywords: long-read sequencing; medical genetics; next-generation sequencing; phasing; pseudogenes; structural variation; tandem repeat expansion.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous