

When and how do patients with cardiac amyloidosis die?
- PMID: 31134330
- PMCID: PMC6952329
- DOI: 10.1007/s00392-019-01490-2
When and how do patients with cardiac amyloidosis die?
Abstract
Background: Cardiac amyloidosis (CA) is an underappreciated cause of morbidity and mortality. Light-chain (AL) and transthyretin (ATTR) amyloidosis have different disease trajectories. No data are available on subtype-specific modes of death (MOD) in patients with CA.
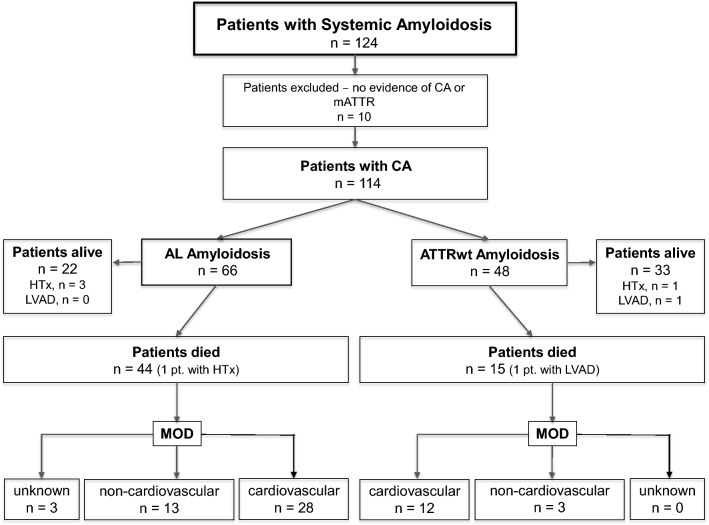
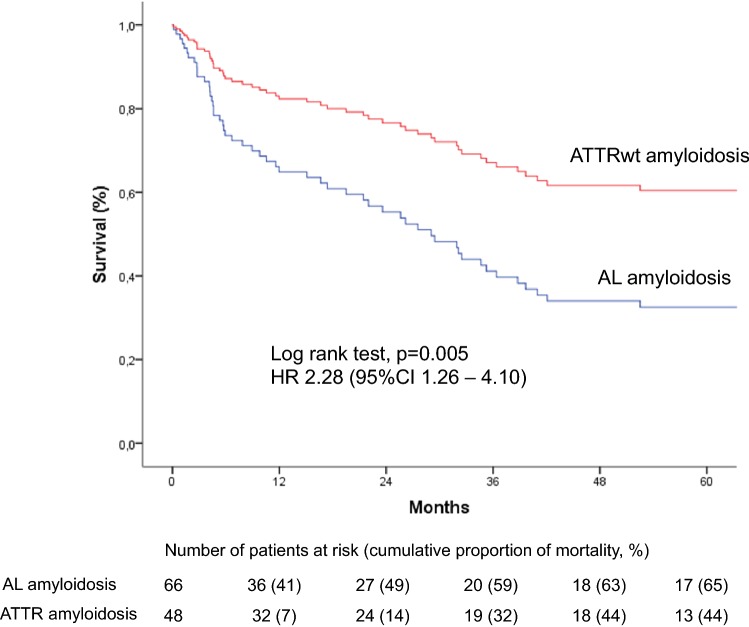
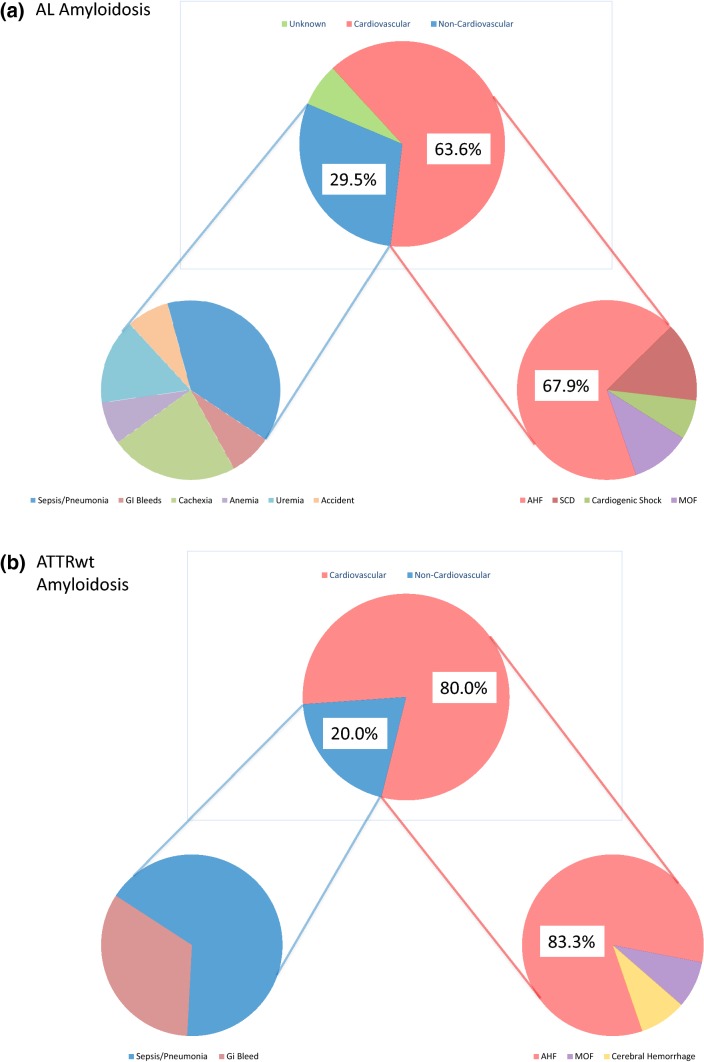
Methods and results: We retrospectively investigated 66 with AL and 48 with wild-type ATTR amyloidosis (ATTRwt) from 2000 to 2018. ATTRwt differed from AL by age (74.6 ± 5.4 years vs. 63 ± 10.8 years), posterior wall thickness (16.8 ± 3.3 mm vs. 14.3 ± 2.2 mm), left ventricular mass index (180.7 ± 63.2 g/m2 vs. 133.5 ± 42.2 g/m2), and the proportions of male gender (91.7% vs. 59.1%), atrial enlargement (92% vs. 68.2%) and atrial fibrillation (50% vs. 12.1%). In AL NYHA Functional Class and proteinuria (72.7% vs. 39.6%) were greater; mean arterial pressure (84.4 ± 13.5 mmHg vs. 90.0 ± 11.3 mmHg) was lower. Unadjusted 5-year mortality rate was 65% in AL-CA vs. 44% in the ATTRwt group. Individuals with AL-CA were 2.28 times ([95%CI 1.27-4.10]; p = 0.006) more likely to die than were individuals with ATTRwt-CA. Information on MOD was available in 56 (94.9%) of 59 deceased patients. MOD was cardiovascular in 40 (66.8%) and non-cardiovascular in 16 (27.1%) patients. Cardiovascular [28 (68.3%) vs. 13 (80%)] death events were distributed equally between AL and ATTRwt (p = 0.51).
Conclusion: Our data indicate no differences in MOD between patients with AL and ATTRwt cardiac amyloidosis despite significant differences in clinical presentation and disease progression. Cardiovascular events account for more than two-thirds of fatal casualties in both groups.
Keywords: Cardiac amyloidosis; Light chain (AL) amyloidosis; Mode of death; Prognosis; Transthyretin (ATTR) amyloidosis.
Conflict of interest statement
Poelzl G has received speaker honoraria from Pfizer and AKCEA Therapeutics.
Figures
References
-
- Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. - DOI - PubMed
-
- Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, Salvi F, Ciliberti P, Pastorelli F, Biagini E, Coccolo F, Cooke RM, Bacchi-Reggiani L, Sangiorgi D, Ferlini A, Cavo M, Zamagni E, Fonte ML, Palladini G, Salinaro F, Musca F, Obici L, Branzi A, Perlini S. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120:1203–1212. doi: 10.1161/CIRCULATIONAHA.108.843334. - DOI - PubMed
-
- Damy T, Costes B, Hagege AA, Donal E, Eicher JC, Slama M, Guellich A, Rappeneau S, Gueffet JP, Logeart D, Plante-Bordeneuve V, Bouvaist H, Huttin O, Mulak G, Dubois-Rande JL, Goossens M, Canoui-Poitrine F, Buxbaum JN. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016;37:1826–1834. doi: 10.1093/eurheartj/ehv583. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
